

表皮生长因子受体(epidermal growth factor receptor,简称 EGFR,又称 ErbB-1 或 HER1)是一种跨膜蛋白,是细胞外蛋白配体表皮生长因子家族(EGF 家族)成员的受体[1]。
EGFR 是 ErbB 受体家族的成员之一,ErbB 受体家族是包含四种密切相关的受体酪氨酸激酶的亚家族:EGFR(ErbB-1)、HER2/c-neu(ErbB-2)、Her3(ErbB-3)以及 Her4(ErbB-4)。影响 EGFR 表达或活性的突变可能导致癌症[2]。

EGFR 与癌症的关系
EGFR 是一种跨膜蛋白,通过结合其特异性配体而被激活。其配体包括表皮生长因子(EGF)和转化生长因子 α (TGF-α)等[3] 。在被其生长因子配体激活后,EGFR 从无活性单体形式转变为活性同型二聚体[4]。除了在配体结合后形成同型二聚体外,EGFR 还可以与 ErbB 受体家族的另一个成员(如 ErbB2/Her2/neu)配对,形成活化的异二聚体[2]。
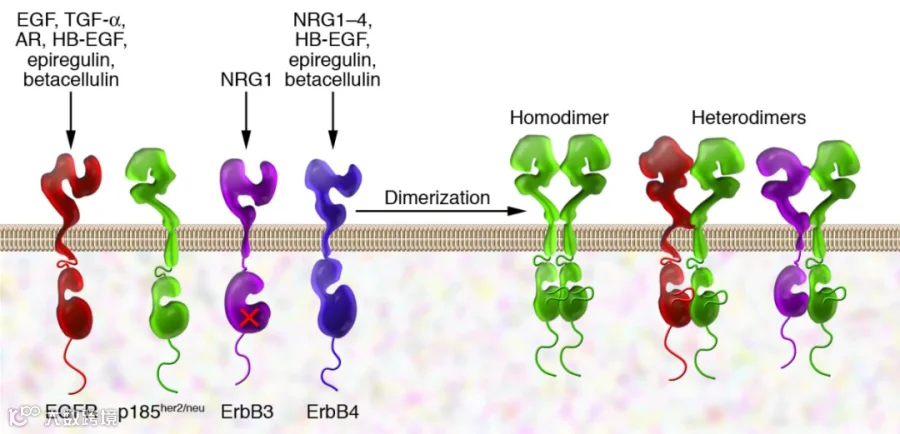
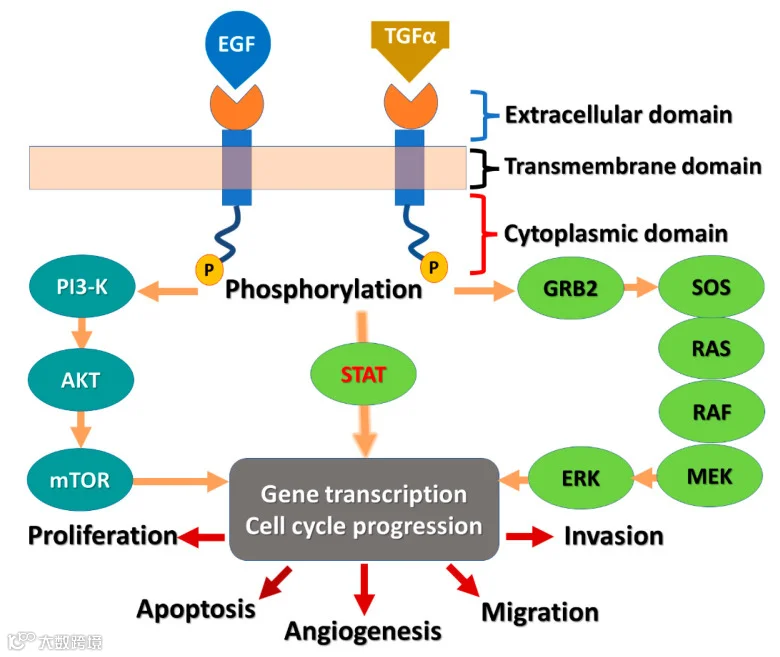
EGFR 二聚化刺激其胞内酪氨酸激酶活性,导致 EGFR 的 C 端结构域中的几个酪氨酸残基(包括 Y992、Y1045、Y1068、Y1148 和 Y1173)发生自磷酸化[5]。这种自磷酸化会引发其他几种蛋白的下游激活和信号转导,这些下游信号转导蛋白启动多个信号转导级联反应,导致 DNA 合成和细胞增殖[6] 。
EGFR 在正常的组织发育和功能中发挥着重要的作用,但是当它们的表达或活性失控时,就会导致细胞增殖过度、抗凋亡、血管生成、侵袭和转移等癌症相关的现象[7] 。
细胞增殖
EGFR 信号通路可以激活 RAS/RAF/MEK/ERK 通路,从而促进细胞周期进程和 DNA 合成。
同时,EGFR 信号通路也可以激活 PI3K/AKT/mTOR 通路,从而促进蛋白质合成和代谢,提供细胞生长所需的能量和物质。此外,EGFR 信号通路还可以激活 JAK/STAT 通路,从而促进一些促进细胞增殖的基因的表达,如 CYCLIN D1、MYC、BCL-XL等。
抗凋亡
EGFR 信号通路可以抑制一些诱导细胞凋亡的信号或分子,如 P53、BAX、BAD 等。
血管生成
EGFR 信号通路可以促进血管内皮细胞的增殖、迁移和分化,从而促进新生血管的形成。同时,EGFR 信号通路也可以促进一些促进血管生成的因子的表达或释放,如 VEGF、FGF、PDGF 等。
侵袭和转移
EGFR 信号通路可以促进癌细胞与周围基质或邻近细胞的附着力和粘着力的降低,从而促进癌细胞的脱落和游离。
同时,EGFR 信号通路也可以促进癌细胞对周围基质或邻近细胞的降解和穿透能力的增强,从而促进癌细胞的侵入和穿越。此外,EGFR 信号通路还可以促进一些促进侵袭和转移的因子的表达或释放,如 MMP、uPA、CXCR4 等。
导致 EGFR 过表达(称为上调或扩增)的突变与许多癌症有关,包括肺腺癌(40% 的病例)、肛门癌[8]、胶质母细胞瘤 (50%) 和头颈上皮瘤 (80-100%)[9] 。这些涉及 EGFR 的体细胞突变导致其持续激活,从而产生不受控制的细胞分裂[10] 。EGFR 或家族成员的突变、扩增或失调与大约 30% 的上皮癌有关[11]。

EGFR 突变类型
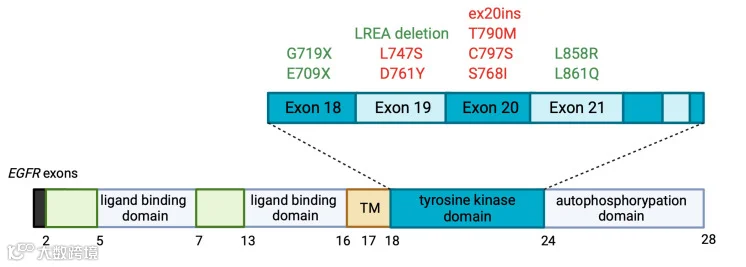
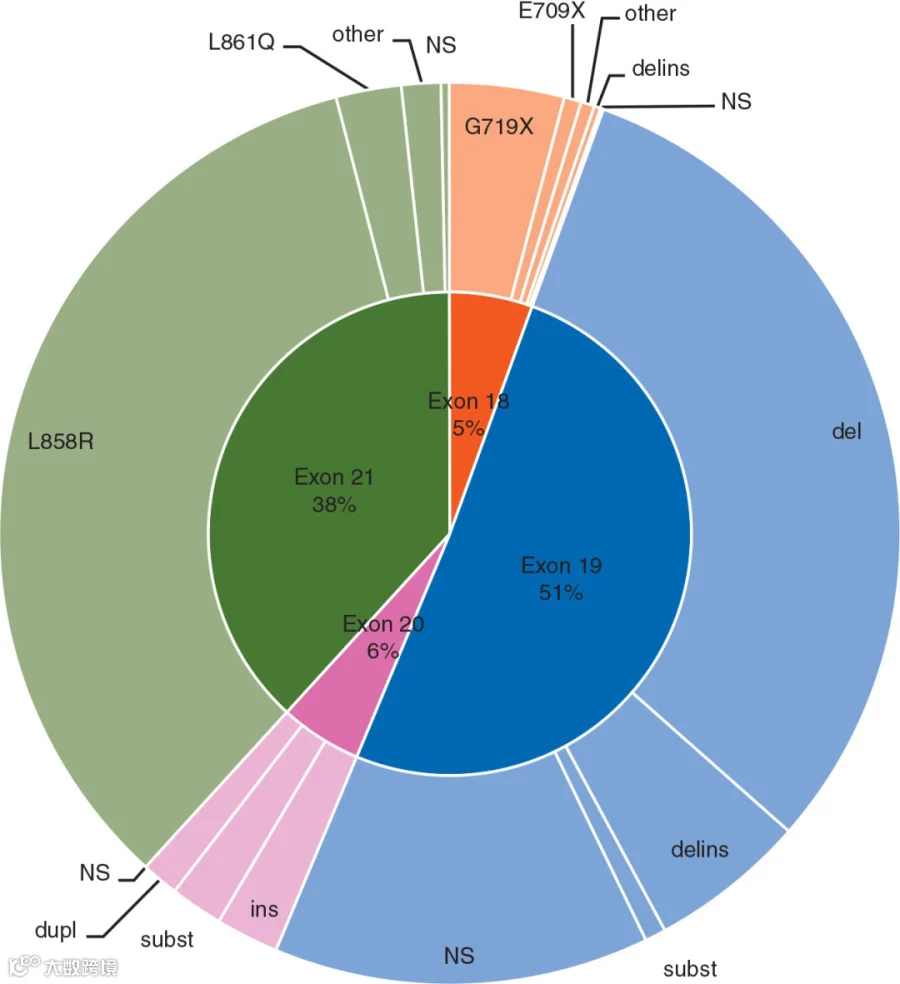
与癌症相关的 EGFR 突变通常发生在其胞内的酪氨酸激酶结构域(外显子 18-24),其中最常见的突变类型是外显子 19 的缺失突变以及外显子 21 的点突变,在非小细胞肺癌(NSCLC)中,这两类突变约占所有 EGFR 突变的 90%[12]。
EGFR 外显子 19 的缺失突变(del747–750),导致 EGFR 酪氨酸激酶结构域中的亮氨酸-精氨酸-谷氨酸-丙氨酸(LREA)基序的缺失。
LREA 缺失会增加 EGFR 自磷酸化并激活下游 AKT 和 STAT 途径,从而促进细胞存活和细胞生长 [13]。外显子 21 的单核苷酸 T>G 突变,导致 858 号密码子的亮氨酸>精氨酸突变(L858R)[14, 15]。
L858R 突变使激酶永久处于活性状态,这导致突变型 EGFR 的活性增加约 50 倍[16]。

针对 EGFR 的靶向药
针对 EGFR 的靶向药物主要分为两类,一类是单克隆抗体(mAb),另一类是小分子酪氨酸激酶抑制剂(TKI)。mAb 与跨膜受体的细胞外结构域结合并阻断其二聚化,TKI 与细胞内结构域的 ATP 结合位点结合,阻断其磷酸化,从而抑制其信号传导[17]。
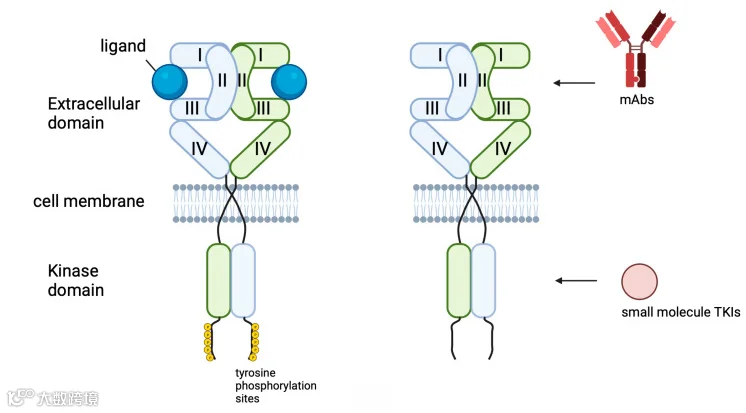
酪氨酸激酶抑制剂(TKI)
酪氨酸激酶(包括 ERBB/HER 受体家族的酪氨酸激酶)的功能是将 ATP 分子的 γ-磷酸基团转移到底物的酪氨酸残基上,从而启动信号进一步传递到下游组分[18]。因此,靶向 EGFR 的酪氨酸激酶活性,可能会消除其在细胞内的信号转导能力。
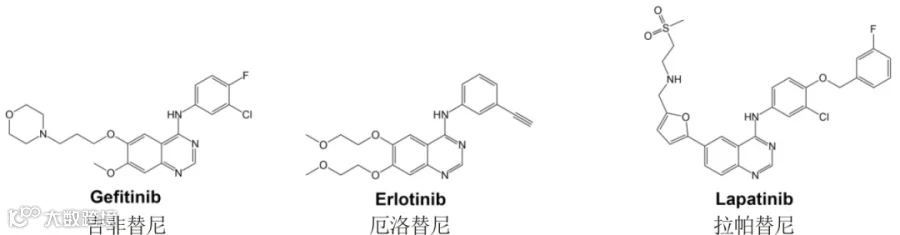
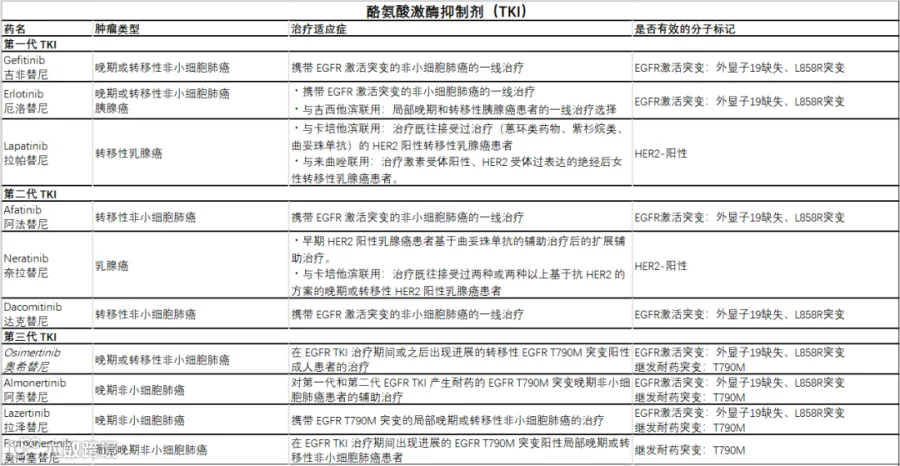
第一代 EGFR 靶向药
吉非替尼(Gefitinib)是一种口服苯胺喹唑啉酮,通过与几个氨基酸残基相互作用,吉非替尼占据 EGFR 酪氨酸激酶结构域中 ATP 结合位点的空间,抑制 EGFR 酪氨酸磷酸化,从而阻断肿瘤细胞中的下游信号级联反应[19]。
2003 年,美国 FDA 批准吉非替尼用于局部晚期或转移性非小细胞肺癌(NSCLC)患者的单药治疗[20]。
2015 年,FDA 批准吉非替尼用于治疗 EGFR 外显子 19 缺失或外显子 21 L858R 替代突变的晚期或转移性 NSCLC 患者[21,22]。
目前,吉非替尼已在 91 个国家获批用于治疗局部晚期或转移性 EGFR NSCLC 成人患者 [21]。
除了吉非替尼之外,第一代 EGFR 靶向药还有厄洛替尼(Erlotinib)以及拉帕替尼(Lapatinib)。
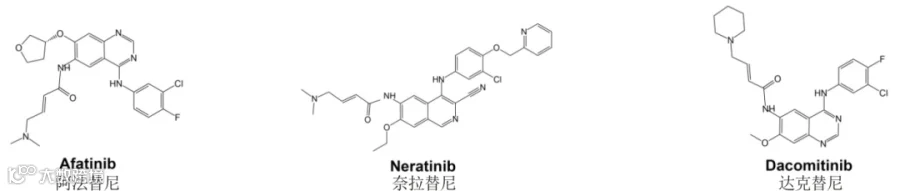
第二代 EGFR 靶向药
对厄洛替尼和吉非替尼的继发性耐药病例的出现迫使研究人员和行业开发新的 EGFR 特异性疗法,以克服其耐药性。
第二代 EGFR TKI 旨在通过抑制额外的伴侣受体酪氨酸激酶(如 HER2)或不可逆地结合激酶结构域来解决获得性耐药问题,从而消除下游 EGFR 信号传导。
阿法替尼(Afatinib)于 2013 年获批用于治疗携带激活性 EGFR 外显子 19 缺失或外显子 21 L858R 替代突变的转移性 NSCLC[23]。
其作用机制与第一代 EGFR 抑制剂厄洛替尼和吉非替尼不同,阿法替尼通过与 ATP 结合位点形成不可逆的共价键,不可逆地抑制 EGFR、HER2 和 HER4 受体的自磷酸化[24]。
它具有独特的丙烯酸酯侧链,与 EGFR C797 残基共价结合,从而导致对 EGFR 酪氨酸激酶的不可逆抑制 [25]。
除了阿法替尼之外,第二代 EGFR 靶向药还有奈拉替尼(Neratinib)以及达克替尼(Dacomitinib)。
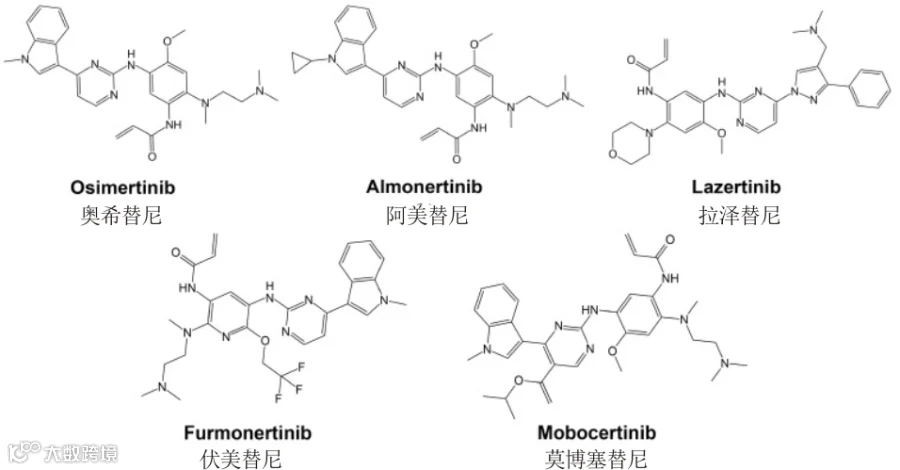
第三代 EGFR 靶向药
第一代低分子量 EGFR 靶向治疗药物厄洛替尼和吉非替尼的缺点是其抑制作用是可逆的,而且它们对继发性 EGFR 突变如 T790M 替代无效。而在超过 50% 的 EGFR 突变型 NSCLC 病例中发现了这种 EGFR 抑制剂引起的耐药突变[26]。为了克服这种频繁的耐药机制,第三代药物被开发出来。
奥希替尼(Osimertinib)是一种口服性 EGFR 特异性 TKI,对晚期 NSCLC 患者的 EGFR 激活突变和继发性 T790M 耐药突变具有很强的选择性[27]。奥希替尼的作用机制是与 EGFR ATP 结合位点的 797-半胱氨酸残基形成共价键[28]。
尽管奥希替尼具有很强的临床疗效,但患者最终也会对这种疗法产生继发性耐药[28]。用奥希替尼治疗 8-10 个月后,可能会出现对该药物的继发性耐药,其中耐药机制可能包括 EGFR C797S 以及 T790M 突变[29]。
除了奥希替尼之外,第三代 EGFR 靶向药还有阿美替尼(Almonertinib)、拉泽替尼(Lazertinib)以及莫博塞替尼(Mobocertinib)等。
第四代 EGFR 靶向药
随着时间的推移,接受第三代 EGFR TKI 治疗的患者对这种治疗产生异质性耐药,包括 EGFR 依赖性的耐药,以及 EGFR 不依赖的耐药[30]。EGFR 依赖性耐药的主要原因是 ATP 结合槽中出现特异性 C797S 点突变[31,32]。另一个原因是继发突变 T790M 引起的缺失突变[32]。
为了克服这些耐药突变,目前正在开发与 EGFR 变构位点结合的第四代药物,现在还处于临床前评估阶段 [33,34]。
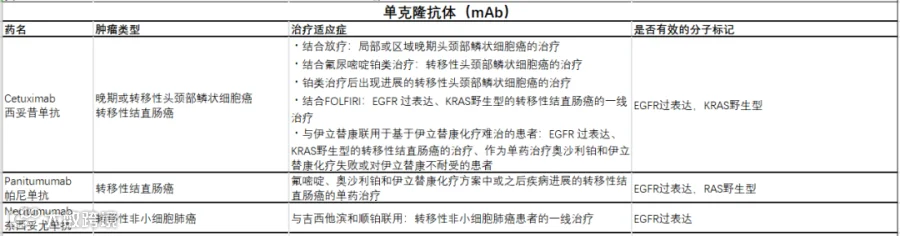
EGFR 特异性单抗
EGFR 特异性单抗通过与 EGFR 结合来破坏促肿瘤生长和存活信号,从而改变其激活状态或阻止配体结合[35]。此外,抗体的特异性结合可以募集免疫细胞来识别和靶向肿瘤细胞[36]。
第二种途径是间接的,通过抗体依赖性细胞毒性(ADCC)机制起作用。例如,自然杀伤细胞(NK 细胞)可以通过 ADCC 机制靶向 mAb 处理的 HER2/neu 过表达细胞[37]。
西妥昔单抗(Cetuximab)是第一个靶向 EGFR 的单克隆抗体,它对人 EGFR 有很强的亲和力,并有效阻断配体的结合,最终导致受体磷酸化和下游信号通路的抑制[38]。
除了竞争性抑制外,西妥昔单抗与 EGFR 的结合还可能诱导受体的内化和降解 [39]。
西妥昔单抗已获得欧洲药品管理局和美国 FDA 的批准,用于局部晚期头颈部鳞状细胞癌(SCCHN)患者,并与伊立替康联合用于治疗转移性结直肠癌(mCRC)。
在美国,西妥昔单抗也被批准用于复发性或转移性 SCCHN 患者以及不能耐受基于伊立替康的方案的 mCRC 患者的单药治疗[40,41]。
除了西妥昔单抗之外,EGFR 特异性单抗还有帕尼单抗(Panitumumab )以及奈西妥尤单抗(Necitumumab)等。
EGFR靶向药汇总

EGFR 靶向药的耐药性
对 EGFR TKIs 的耐药机制多种多样,包括 MET 扩增、KRAS 突变、BRAF 突变、PIK3CA 突变、SCLC 转化、PTEN 缺失等等[42]。总的来说,主要可以分为四种类型[43]。
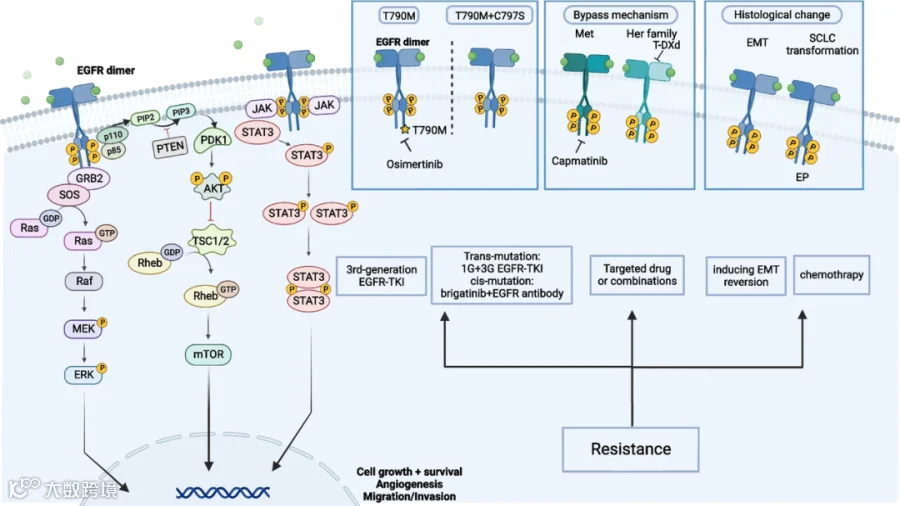
1
EGFR 依赖型耐药
此类耐药因 EGFR 本身的改变而引起,这些改变可以影响 EGFR 与药物的结合或信号传导,从而使药物失效。
例如非小细胞肺癌(NSCLC)中经常发生的 T790M 突变,这是一种导致 EGFR 激酶结构域中 ATP 结合位点上第 790 位氨基酸由苏氨酸变为甲硫氨酸的突变,它可以增加 EGFR 对 ATP 的亲和力,从而降低吉非替尼和厄洛替尼等 TKI 的结合能力和抑制效果[26]。
又如 EGFR C797S 突变,它是对奥希替尼耐药的最常见机制之一。在 EGFR C797S 突变中,奥希替尼不能与突变型 EGFR 共价结合,从而失去其疗效[29]。
2
旁路途径激活突变引起的耐药
细胞信号转导具有冗余机制,同一条信号通路可以被一种受体与其配体结合而激活,也可能被另一种受体与其配体结合而激活,这就为肿瘤细胞绕过药物的抑制提供了基础。
在大约 20% 的 EGFR TKI 耐药病例中观察到 MET 基因的扩增。MET 通过与其配体肝细胞生长因子(HGF)结合,激活 PI3K/AKT/mTOR 信号通路,绕过 TKI 对 EGFR 信号通路的抑制[44]。
3
EGFR 下游信号通路激活突变引起的耐药
EGFR 下游信号通路的改变可以影响 EGFR 信号通路的活性或平衡,从而使药物失效。
例如,结直肠癌中经常发生的 KRAS G12V 突变,这是一种导致 KRAS 蛋白中第 12 位氨基酸由甘氨酸变为缬氨酸的突变,它可以使 KRAS 蛋白处于持续激活状态,从而激活 RAS/RAF/MEK/ERK 通路,绕过赛可瑞等单克隆抗体对 EGFR 信号通路的抑制[45]。
4
组织学转化引起的耐药
最近的一项研究表明,组织学变化可能与获得 EGFR TKI 耐药有关[46]。上皮表型缺失和间充质特征增加的过程称为上皮间质转化(EMT),EMT 与肿瘤细胞迁移和侵袭能力增加有关。EMT 在肿瘤耐药中的作用是一个新兴的研究领域,更多的细节仍待研究发现。

克服 EGFR 靶向药耐药性的策略
尽管针对不同类型癌症的靶向药物应用在世界范围内取得了明显的成功,但仍存在尚未得到充分解决的挑战。每一种下一代 EGFR 靶向药物都是为了克服肿瘤对上一代药物的耐药性,或覆盖上一代药物最初不起作用的患者[47]。
然而,当这些药物被设计为靶向具有特定突变的肿瘤时,它们对野生型 EGFR 不再有效,从而导致它们在大量患者群体中被放弃使用。
此外,对第三代药物可能产生继发性耐药,例如,EGFR 酪氨酸激酶结构域的 ATP 结合位点 C797S 的密码子 797 处的丝氨酸取代半胱氨酸 [48]。
这已经迫使研究人员使用组合策略并测试第四代 EGFR 靶向的候选分子。这种持续的动态类似于军备竞赛,突变出现,靶向疗法被开发和应用,作为回应,EGFR 的后续继发突变出现在肿瘤克隆中。
患者的获益时间可能从几个月到几年不等;然而,不幸的是,肿瘤不可避免地会对所使用的靶向药物产生耐药性。
因此,除了寻求靶向耐药突变的新型药物外,联合治疗方案似乎是一条合理的路线。这种方法涉及将具有不同特异性的多种药物与常规化疗相结合。
例如,联合使用单抗和 TKI,可以同时抑制 EGFR 的胞外和胞内信号传导,从而增强对 EGFR 信号通路的抑制;联合使用针对 EGFR 和其他信号通路的药物,可以同时抑制 EGFR 信号通路和其下游或交叉激活的信号通路,从而降低对 EGFR 信号通路的依赖性或冗余性。
此外,探索导致对 EGFR TKI 耐药的潜在细胞和分子机制并利用个性化预测方法可以提高 EGFR 靶向治疗的效果。例如,检测癌细胞中 EGFR 基因是否存在 T790M 突变,可以评估患者对吉非替尼等小分子 TKI 是否具有耐药性。
参考文献(上下滑动):
点击下方名片,关注我们