


1.1
致病假说的演进:从级联反应到多因素网络
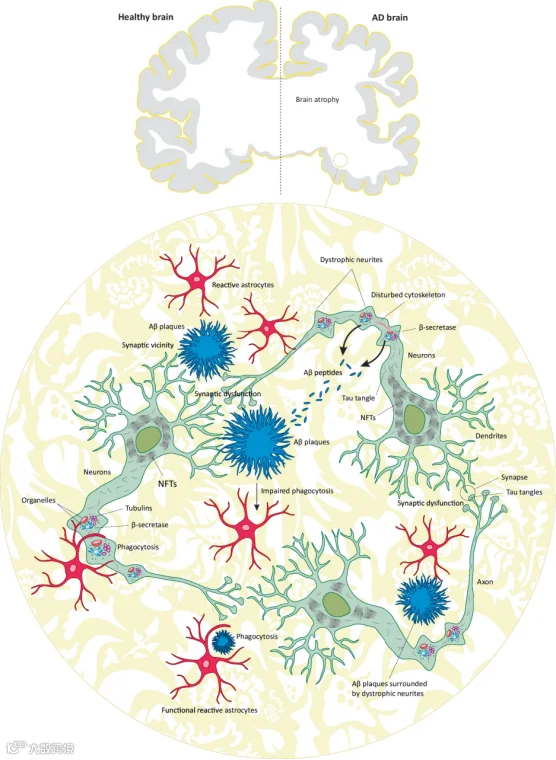
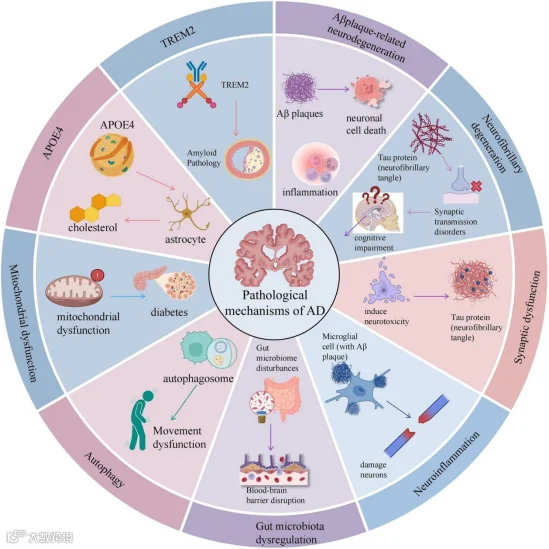
淀粉样级联假说:该假说认为Aβ的生成与清除失衡是AD的始动因素。其最强有力的证据来自家族性AD的遗传学研究——APP、PSEN1基因突变直接导致Aβ42过度生成。这直接催生了以5xFAD、3xTg-AD为代表的经典转基因小鼠模型,这些模型极大地推动了我们对Aβ病理机制的理解,并成为药物开发的主导理论。然而,针对Aβ的临床试验屡遭挫折,提示单纯清除Aβ难以逆转已建立的病理网络,疾病可能存在更复杂的下游机制[ 3-5]。
- Tau蛋白假说:研究发现,NFTs的空间分布与认知衰退程度高度相关,其播散模式(Braak分期)与疾病进展阶段惊人地一致。近年来,研究焦点从终末的NFTs转向了更具神经毒性的可溶性Tau寡聚体,并揭示了Tau蛋白可通过“朊蛋白样”机制沿神经环路传播,成为疾病进展的关键驱动因素[2]。此外,Tau的多种翻译后修饰,如磷酸化、乙酰化、泛素化等,共同构成了复杂的“Tau PTM谱”,其中特定的修饰组合(如p-tau217、p-tau231)已成为早期诊断和追踪疾病进展的核心生物标志物[6,7]。
神经炎症假说:近年来,神经炎症被认为是AD进展的核心驱动力。全基因组关联分析(GWAS)发现,TREM2、CD33等免疫相关基因的突变显著改变AD患病风险[2, 3]。小胶质细胞和星形胶质细胞在AD中呈现“双刃剑”效应:早期可能起清除病理蛋白的保护作用,晚期则转为慢性炎症损伤。更重要的是,小胶质细胞在Aβ与Tau的协同中扮演“中介”角色——Aβ激活的小胶质细胞可通过胞外囊泡将Tau传递至邻近神经元,加速病理播散[2,3]。
突触与环路损伤假说:突触功能障碍和神经环路失连接是认知下降的直接结构基础。Aβ寡聚体(AβOs)和Tau寡聚体均可在突触水平直接损伤长时程增强(LTP),且二者具有协同毒性。突触丢失的严重程度是预测认知功能下降最相关的病理指标。最近的研究表明,Tau寡聚体甚至在NFT形成之前就富集于突触,直接破坏突触功能和结构[6,8]。
线粒体功能障碍与自噬障碍:线粒体功能障碍导致能量代谢衰竭和氧化应激,而自噬-溶酶体系统功能障碍则导致异常蛋白(如Aβ和Tau)的清除受阻,两者共同构成AD病理的恶性循环[9]。
2.1
传统动物模型的贡献与局限
传统动物模型在AD的研究史上发挥了不可替代的奠基作用,推动了从病理机制解析到药物筛选的多个关键领域:
-
化学诱导模型:通过药物(如东莨菪碱)或毒素损伤胆碱能系统,能快速模拟认知障碍,但无法复刻Aβ斑块和NFTs这两个核心病理[10,11]。
-
转基因小鼠模型的“黄金时代”:家族性AD基因突变的发现,催生了转基因小鼠的“黄金时代” [12]。从最早的PDAPP小鼠[13]、Tg2576小鼠[14],到同时携带APP、PS1和Tau突变的3xTg-AD小鼠[15],再到病理进展迅速的5xFAD小鼠[16],这些模型极大地推动了我们对Aβ和Tau病理机制的理解。
-
敲入模型的兴起:为了克服传统转基因模型过度表达和人源基因的局限,研究者开发了敲入(Knock-in)小鼠,如AppNL-G-F,在保持内源性表达水平的同时引入人源化FAD突变。这类模型更接近生理状态,但通常仍缺乏tau病理和显著神经元丢失[17]。
-
多样化模型生物的拓展:研究者还利用大鼠(如TgF344-AD,该模型同时出现Aβ斑块、Tau病理、神经元丢失和年龄依赖性认知障碍,比多数小鼠模型更接近人类AD进程)、斑马鱼、果蝇、线虫乃至非人灵长类(如转基因狨猴)来研究AD的不同维度。这些模型在遗传筛选和药物筛选中具有独特优势[18,19]。
周期长、成本高:构建和繁殖转基因品系需要数月至数年。
难以模拟散发性AD:绝大多数AD患者为散发性,缺乏明确的单一基因突变,且受APOE4等风险基因和环境因素共同影响。
无法精确控制时空表达:病理蛋白在发育早期即开始全身性表达,难以区分其在发育和成年期的作用,也无法精准研究特定脑区病理变化对环路的影响。
遗传背景固定:难以在特定遗传背景上灵活引入新的病理改变(如APOE4敲入、TREM2突变)。
种属差异:小鼠的Aβ和Tau蛋白与人类存在序列和翻译后修饰上的差异,这可能解释了为何许多在转基因小鼠中有效的药物在临床试验中失败[20]。
2.2
AAV技术:一种“按需定制”的灵活建模策略
区域精准(Delivery):通过立体定向注射,将AD相关基因(如APP、Tau、APOE4)精准递送至目标脑区(如海马、内嗅皮层),模拟AD病理发生的区域特异性,尤其适用于研究病理沿神经环路的播散机制。不同AAV血清型(如AAV1/2、AAV8、AAV9、AAVrh.10)在脑内具有不同的转导效率和细胞嗜性,可根据研究目的进行选择[21-23]。
时间精准(Duration):可在动物的任何年龄段(幼年、成年或老年)进行病毒注射,在特定时间点启动病理过程,为研究疾病动态进展和干预时间窗提供了可能。
模型定制精准(Design):一方面,可通过在同一动物体内同时递送多个基因(如APP+Tau),快速构建复合病理模型,探索不同假说之间的协同作用;另一方面,可在任意遗传背景的小鼠(如野生型、APOE4敲入鼠、TREM2突变鼠)上按需建模,精准模拟遗传风险与环境因素的交互作用。
3.1
验证淀粉样级联假说:聚焦Aβ生成失衡与早期突触损伤
3.2
连接淀粉样级联与Tau假说:揭示Aβ与Tau的协同作用
3.3
模拟散发性AD与神经炎症假说:解析APOE4与TREM2的作用
3.4
AAV介导的Tau传播模型:解析病理蛋白的时空动态
-
Tau的传播依赖于年龄:老年小鼠(22-24月龄)中P301Ltau的传播效率是年轻小鼠(3月龄)的2倍以上,为AD发病与年龄的强相关性提供了直接的实验证据。 -
传播与聚集可解耦:可溶性非聚集型Tau同样能够在神经元间传播,且Tau在供体神经元中即使发生错误折叠,也不一定进展为成熟的NFT,提示存在一个潜在的治疗窗口期。 -
脑区特异性差异:同一AAV-P301Ltau注射至内嗅皮层可诱导Tau错误折叠和传播,但注射至纹状体则无此效应,揭示了不同脑区神经元对Tau病理的内在易感性差异。
-
递送抗Tau抗体或Tau降解酶:利用AAV在中枢神经系统持续表达针对特定Tau表位的抗体(如临床在研的Etalanetug[34]),或表达Tau蛋白特异性降解酶,实现长效的Tau清除。这种策略可以避免反复给药带来的免疫原性问题,并能将治疗性蛋白精准递送至脑内。
-
递送抗炎因子与调节免疫细胞:通过AAV递送IL-10、TGF-β等抗炎细胞因子,或将TREM2等关键免疫调节分子过表达于小胶质细胞,旨在将神经炎症从有害的慢性激活状态“重编程”为有益的保护性状态[32]。 -
CRISPR基因编辑:利用AAV递送CRISPR-Cas9系统,对APOE4等风险基因进行原位编辑,将其转换为保护性亚型(如APOE2),从根源上干预遗传风险。这种策略在散发性AD,特别是APOE4携带者中,具有巨大的治疗潜力。Gyorgy等人[37]已使用该技术在Tg2576小鼠中实现了对瑞典突变APP等位基因的特异性编辑,为家族性AD的基因治疗提供了概念验证。
-
多靶点联合治疗:随着抗Aβ单抗(如lecanemab)和抗Tau疗法(如反义寡核苷酸BIIB080[35, 36])的临床进展,AAV模型可作为理想的平台,用于模拟和优化联合治疗的时序与协同效应。例如,构建同时表达APP突变和Tau突变的AAV模型,用于评估Aβ靶向药与Tau靶向药联用的最佳时机和效果。一个AAV载体同时递送两种治疗性基因(如抗炎因子+抗Tau抗体)也是未来极具前景的方向。
免疫原性:高剂量AAV注射本身可能引发局部炎症反应,与AD的神经炎症研究相互干扰。
表达的异质性:病毒注射的范围和感染效率在不同动物个体间可能存在差异,影响病理程度的均一性。
治疗的安全性:作为基因治疗载体,AAV的长期表达安全性、潜在的基因组整合风险及免疫原性仍需深入评估。此外,如何安全地靶向Tau而不干扰其生理功能(如微管稳定)是核心挑战之一 [4]。
系统性问题:局部注射难以完全模拟AD作为全脑性疾病的复杂性,而系统性给药则面临血脑屏障的阻碍和肝脏富集的问题。新近开发的AAV9及其变体(如AAV.PHP.eB)可有效穿越血脑屏障,为全脑递送提供了新可能[27]。
破解病理传播:结合光遗传学与AAV,精确操控特定环路,实时动态研究Aβ和Tau蛋白的“朊蛋白样”传播机制,并利用AAV递送针对特Tau的抗体,实现精准阻断。
构建“人性化”网络模型:在携带人源化风险基因(如APOE4、TREM2-R47H)的小鼠上,叠加AAV诱导的病理,精准模拟遗传-病理-炎症多因素交织的散发性AD网络。
加速联合治疗策略评估:利用AAV模型模拟抗Aβ与抗Tau疗法的联合应用,探索协同效应。同时,AAV介导的“单次给药、长期有效”的多靶点联合基因治疗(如同时递送抗炎因子和抗Tau抗体)将是未来探索的重要方向[6]。
推动临床转化:结合先进的生物标志物(如脑脊液中的MTBR-tau243、血浆中的p-tau217)和成像技术(如tau PET),AAV模型可为新一代Tau靶向疗法的剂量选择、给药时机和疗效评估提供关键的临床前依据。
AAV在AD研究中的应用方案总结:目前APP过表达多采用转基因/敲入小鼠模型,而Tau过表达可采用AAV立体定位注射实现快速建模。血清型方面,海马局部注射常用AAV1、AAV1/2、AAV8、AAV9、AAVrh.10,均能高效感染海马神经元;若需全脑递送,可选择AAV.PHP.eB通过尾静脉注射,可实现全脑神经元广泛转导。启动子选择需根据靶细胞类型灵活调整:神经元过表达常用CAG、CBA或hSyn;星形胶质细胞靶向可用GFAP启动子;若研究小胶质细胞,则可选用CD68或F4/80启动子实现小胶质细胞特异性表达。注射方式以立体定位注射为主,全脑递送则采用静脉注射。剂量文献报道跨度较大(局部注射1×10⁸~1×10¹¹ GC/位点,静脉注射约1×10¹²~1×10¹⁴ GC/kg),且不同病毒滴度、注射操作和动物品系均会影响转导效率,因此建议设置剂量梯度进行预实验,确定适合自身实验室条件的最佳剂量。


|
|
|
|
|
|
|
|
|
|
|
|

END


长按二维码
关注我们吧
