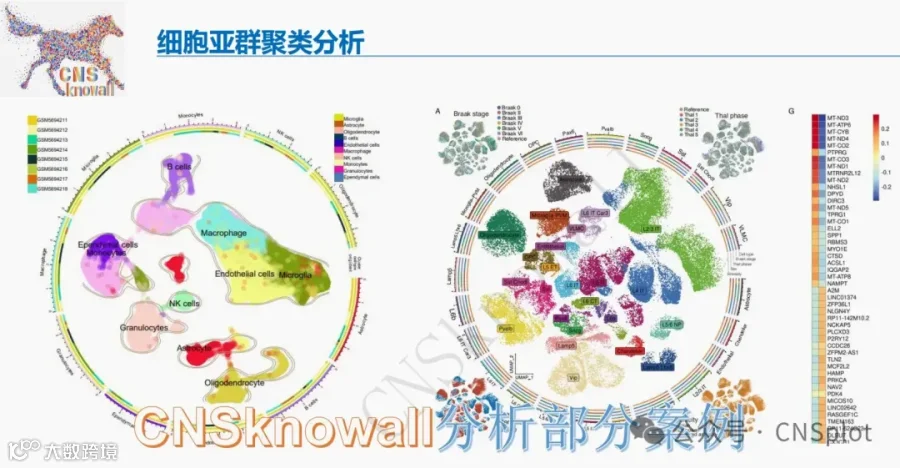
今天给大家解读一篇3月发表在《Stem Cell Research & Therapy》上的题目为“Targeting p75NTR activity alleviates the neurotoxic effect of high glucose on iPSC-derived dopaminergic neurons.”的文章。本研究旨在探究高糖损害多巴胺能神经元的机制。通过使用人iPSC来源的细胞模型,发现高糖激活pro-NGF/p75NTR轴直接导致神经元凋亡,并同时激活胶质细胞释放神经毒性因子。抑制p75NTR可有效挽救神经元,提示该受体是关键的干预靶点。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!包括数据集、绘图代码、图表复现、思路总结、参考文献……0代码!鼠标点点点即可轻松完成5-10分生信SCI全文复现!
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!








团队成员合影(位于上海陆家嘴中心,可随时预约参观)






题目:《靶向p75NTR活性可缓解高糖对iPSC来源的多巴胺能神经元的神经毒性作用》Targeting p75NTR activity alleviates the neurotoxic effect of high glucose on iPSC-derived dopaminergic neurons
发表期刊:Stem Cell Research & Therapy
影响因子:7.3
研究背景:
高血糖是糖尿病的特征,严重影响神经系统。流行病学和动物研究证据表明糖尿病与多巴胺能功能障碍及帕金森病风险增加有关,但机制不明。鉴于神经营养因子在糖尿病神经系统表现中的作用,本研究聚焦p75NTR受体在高糖条件下多巴胺能神经退行性变中的角色。
研究思路:
- 建立模型
用高糖(50mM, 100mM)处理iPSC来源的多巴胺能神经元、星形胶质细胞和小胶质细胞48小时,模拟高血糖。 - 评估毒性
通过细胞毒性检测、RNA测序和DNA损伤评估,研究高糖对神经元的病理改变。 - 靶向验证
药理学靶向p75NTR活性,探究其在葡萄糖神经毒性中的作用。 - 胶质细胞作用
利用条件培养基和炎症标志物分析,评估胶质细胞介导的神经毒性。
研究亮点:
- 模型优势
采用人iPSC分化的多巴胺能神经元、星形胶质细胞和小胶质细胞,更贴近人类病理生理。 - 机制深入
不仅阐明了神经元内在的p75NTR依赖的凋亡通路,还揭示了胶质细胞介导的间接神经毒性作用。 - 转化潜力
明确了p75NTR作为核心治疗靶点,并验证了其抑制剂及模拟物BNN27的神经保护作用,为干预提供了直接证据。
研究结果:
-
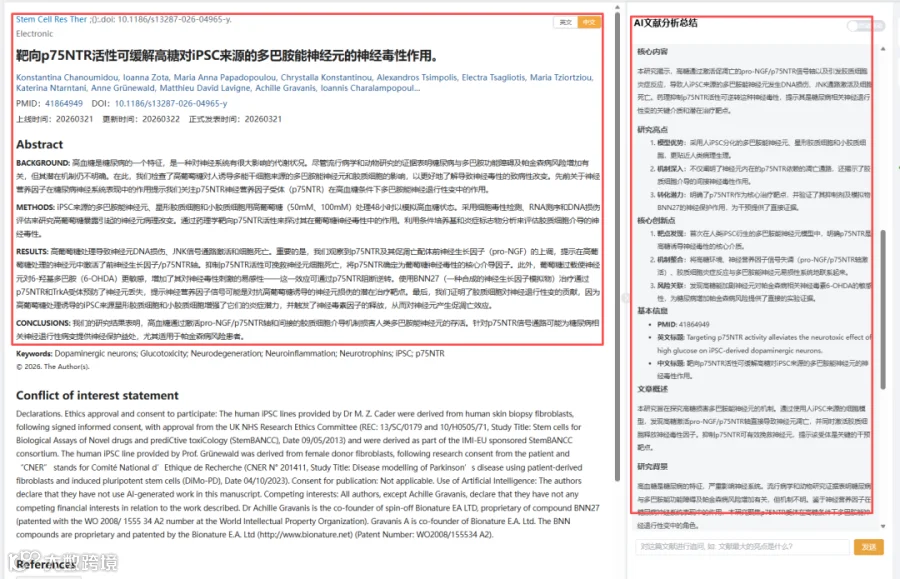
高糖处理导致神经元DNA损伤、JNK信号激活和细胞死亡。 -
高糖神经元中p75NTR及其促凋亡配体pro-NGF表达上调,表明pro-NGF/p75NTR轴被激活。 -
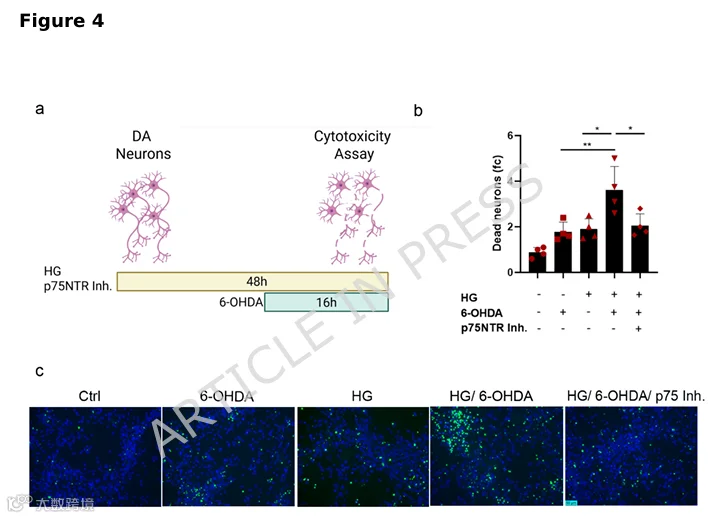
抑制p75NTR活性可挽救神经元死亡,证实p75NTR是葡萄糖神经毒性的核心介质。 -
高糖使神经元对神经毒素6-OHDA更敏感,此效应可被p75NTR阻断逆转。 -
合成NGF模拟物BNN27通过p75NTR和TrkA受体可预防神经元丢失。 -
高糖处理的胶质细胞炎症潜能增强,释放神经毒性因子,对神经元产生促凋亡效应。
研究总结:
结论:高糖通过激活pro-NGF/p75NTR轴以及间接的胶质细胞介导机制,损害人多巴胺能神经元的存活。靶向p75NTR信号通路可能为糖尿病相关的神经退行性变,特别是针对有帕金森病风险的患者,提供神经保护益处。
讨论:研究结果将高血糖、p75NTR信号异常激活与多巴胺能神经元特异性损伤直接关联,为理解糖尿病中枢神经系统并发症的分子机制提供了新视角。所发现的p75NTR依赖机制是一个明确的药物干预窗口,BNN27等药物的有效性为后续治疗策略开发奠定了基础。同时,研究强调了在糖尿病神经病变中,神经元-胶质细胞互斥的重要作用。
结果译文:
1.高糖诱导DA神经元DNA损伤及细胞死亡

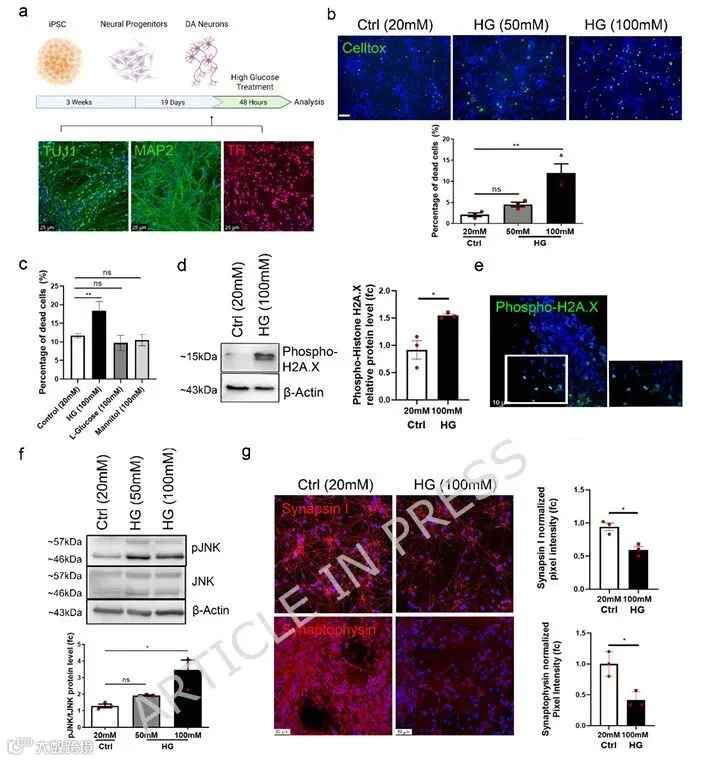
2.高糖处理神经元的全转录组分析显示应激相关过程诱导
为更深入了解高糖对DA神经元的分子影响,我们进行RNA测序以比较HG处理与对照神经元的全基因表达谱。葡萄糖处理诱导神经元转录变化,与非处理细胞相比,81个基因上调,209个基因下调(p adj < 0.05)(图2a,b,附加文件1:图S1,附加文件2:表S1,附加文件3:表S2)。对上调基因的基因本体论(GO)和Reactome基因集(RGS)富集分析表明,与细胞应激相关的过程被诱导,包括DNA损伤修复、分泌负调控及细胞衰老(图2c,d,附加文件4:表S3)。值得注意的是,参与胆固醇生物合成的基因如MVK和FDFT1上调,与先前将高糖与增强胆固醇生成相联系的研究一致(图2b,d)。有趣的是,最显著富集的GO term是“细胞周期时相转换”,尽管DA神经元为有丝分裂后。这提示HG触发细胞生存和周期调控基因的代偿性表达,包括BIRC5(染色体乘客复合体组分)和CCNB1/2(附加文件3:表S2,附加文件4:表S3,附加文件6:表S5)。与HG处理神经元中观察到的DNA损伤诱导一致,通路分析显示PLK1被诱导。PLK1是DNA损伤应答(DDR)机制的一部分,保护细胞免于携带受损DNA进入细胞周期(图2e)。此外,促凋亡通路包括p53和p73被诱导(图2e)。同时,对下调基因的分析表明突触后细胞骨架组织发生变化,并影响与DA神经元神经保护相关的ERA基因组通路(图2c,e,附加文件5:表S4,附加文件6:表S5)。总体而言,RNA测序分析证实了HG下DNA损伤和细胞死亡诱导的实验证据。
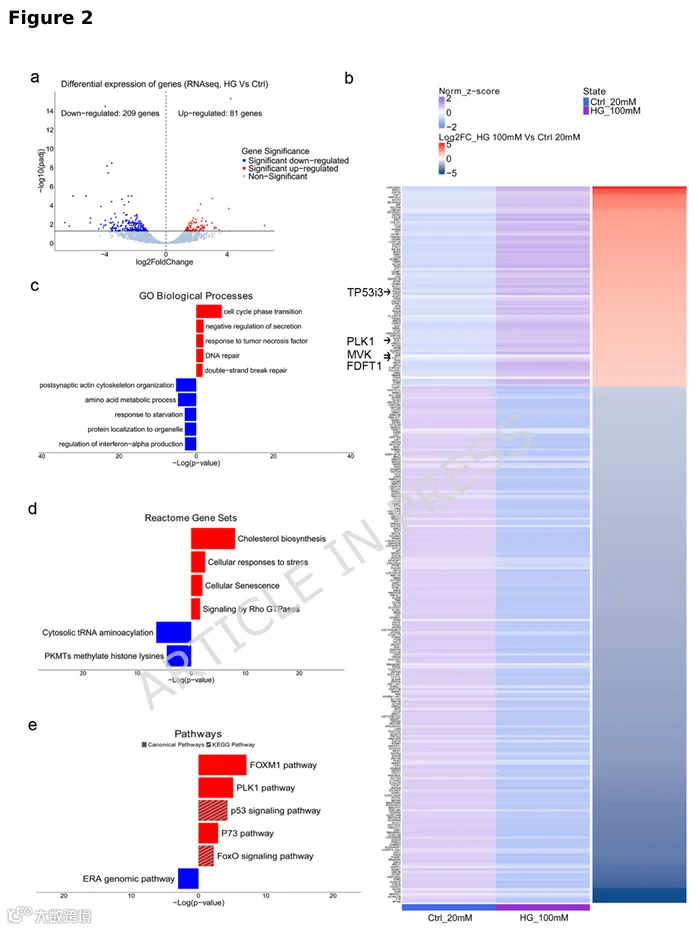
3.p75NTR上调并参与高糖驱动的DA神经元细胞死亡
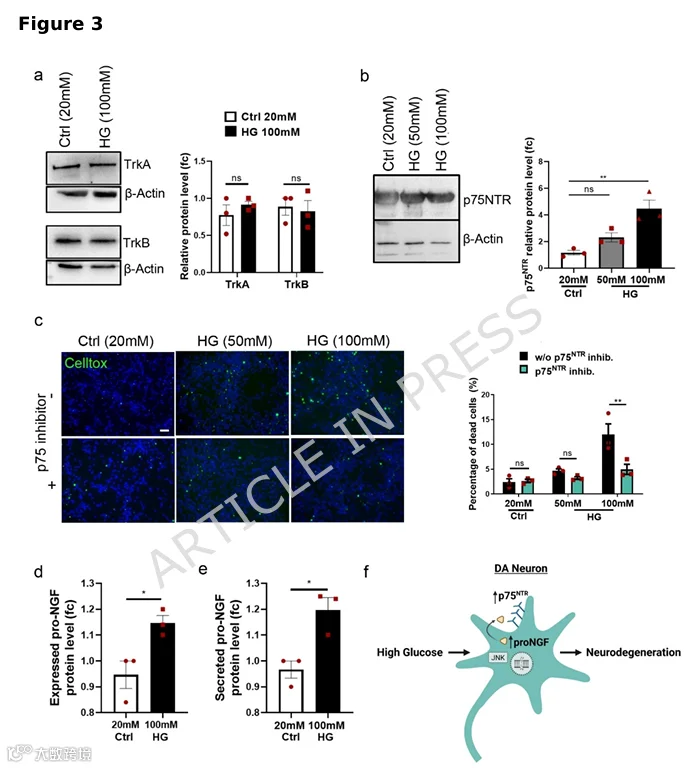
4.高糖增加神经元对6-OHDA毒性的易感性,该效应被p75NTR抑制所减弱
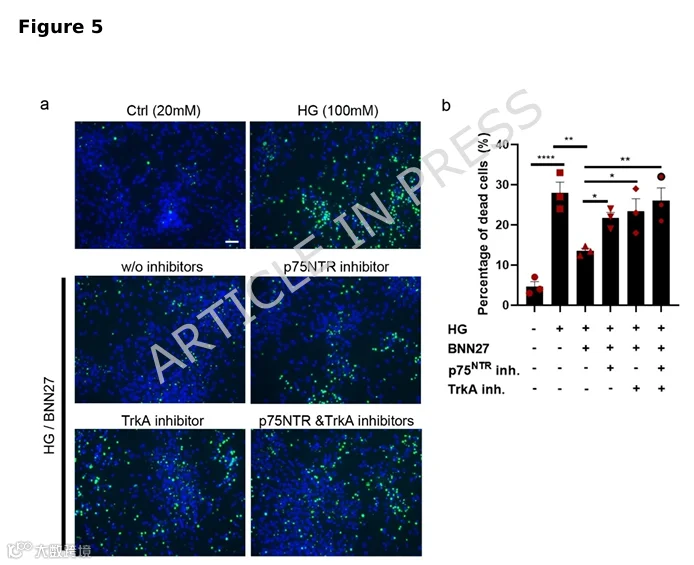
5.BNN27通过p75NTR和TrkA受体在DA神经元中发挥神经保护作用
基于p75NTR的促凋亡功能,我们接下来探究神经营养因子信号是否可作为治疗靶点以减轻HG驱动的神经退行性变。我们测试了一种合成小分子17-螺甾类固醇类似物BNN27,其作为NGF模拟物,已在糖尿病视网膜病变和阿尔茨海默病中作为神经保护治疗分子引起关注。我们及其他课题组先前发表的研究表明,BNN27作为NGF受体TrkA和p75NTR受体的选择性激活剂,促进多种类型神经元的生存。多巴胺能神经元暴露于1μM BNN27显著减轻高糖诱导的细胞死亡。此保护效应通过抑制p75NTR或TrkA(单独或联合)而被消除,表明BNN27通过两种受体发挥其保护作用(图5a,b)。总之,我们的发现强调BNN27是一种有前景的基于神经营养因子的候选治疗分子,能够减轻HG诱导的多巴胺能神经退行性变。
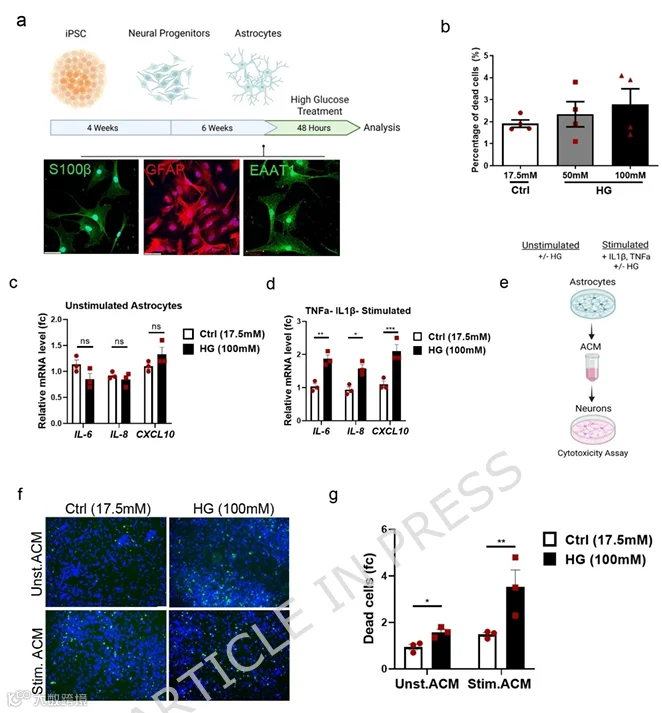
6.高糖增强星形胶质细胞炎症潜力并诱导神经毒性因子分泌
文献当前共识认为,神经炎症是DM与神经退行性疾病之间的关键联系。星形胶质细胞是大脑中主要的稳态细胞,但在应激刺激下,它们表现出表型变化,可能增加神经退行性变风险。在此背景下,我们探究了人星形胶质细胞在高糖条件下对多巴胺能神经退行性变的作用。我们如先前所述将人iPSCs分化为星形胶质细胞。通过星形胶质细胞标志物GFAP、S100β和EAAT1的免疫染色鉴定细胞身份(图6a)。成熟星形胶质细胞用HG(100mM D-葡萄糖)处理48h以模拟高糖。细胞生存分析显示HG对星形胶质细胞无凋亡效应,表明星形胶质细胞比神经元对HG更具抵抗力(图6b)。为探讨HG对星形胶质细胞活化的影响,我们分析了在存在或不存在外源性促炎刺激条件下炎症标志物的表达。星形胶质细胞用HG和/或30ng/ml TNFα和10ng/ml IL-1β处理以诱导活化。qPCR分析显示,在HG处理的刺激条件下而非未刺激星形胶质细胞中,IL-6、IL-8和Cxcl10基因表达增加(图6c,d),暗示HG本身不足以激活这些基因,但加剧星形胶质细胞对促炎刺激的反应性。最后,为评估HG处理的星形胶质细胞如何影响神经元生存,我们收集了细胞因子刺激和未刺激星形胶质细胞的星形胶质细胞条件培养基(ACM)。DA神经元暴露于ACM 48h,不额外添加葡萄糖(图6e)。值得注意的是,来自高糖星形胶质细胞(无论有无外源性细胞因子刺激)的ACM均引起显著神经元死亡,细胞因子刺激组效应更强。具体而言,HG处理星形胶质细胞的ACM使神经元死亡较对照条件增加1.6倍,而细胞因子和HG处理星形胶质细胞的ACM导致2.3倍增加(图6f,g)。尽管未刺激细胞中细胞因子表达水平未改变,但其他具有毒性特性的分泌因子可能解释此效应。结论性地,我们的结果表明HG增强星形胶质细胞的炎症潜力,并诱导对DA神经元具有有害效应的神经毒性因子的分泌。

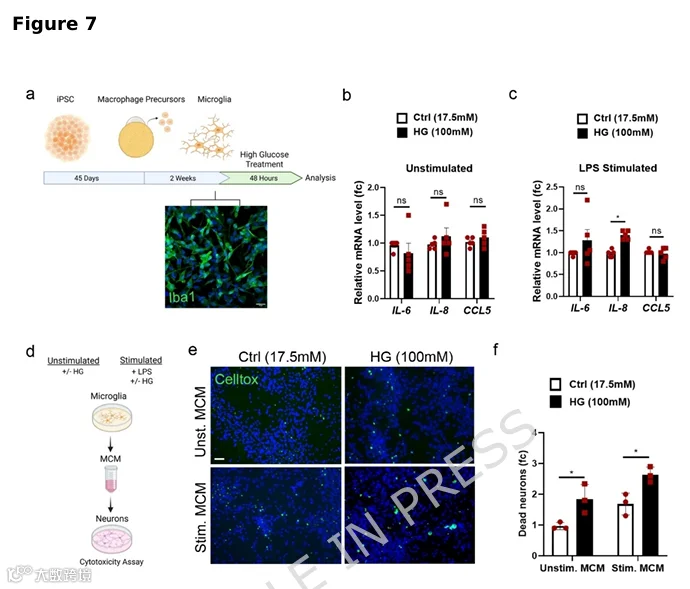
7.高糖导致小胶质细胞神经毒性反应
小胶质细胞是CNS的常驻巨噬细胞,是大脑神经炎症的主要介质。活化的小胶质细胞可能通过释放细胞因子、趋化因子和谷氨酸损害神经元活动。为探究高糖对人小胶质细胞的影响及其对DA神经病理学的继发效应,我们如先前所述从人iPSCs生成Iba1+小胶质细胞(图7a)。小胶质细胞暴露于100mM D-葡萄糖48h,并在最后6h给予或不给予100ng/ml LPS以模拟促炎微环境。与星形胶质细胞相似,IL-6、IL-8和Ccl5的qPCR分析显示,小胶质细胞暴露于HG不诱导这些炎症标志物表达(图7b),尽管LPS刺激在HG下增强IL-8表达(图7c)。我们接下来收集小胶质细胞条件培养基(MCM)用于神经元处理(图7d)。来自HG处理小胶质细胞(无论有无LPS刺激)的MCM增加神经元细胞死亡。与对照相比,暴露于高糖未刺激和LPS刺激小胶质细胞MCM后,死亡神经元比例分别增加1.9倍和1.5倍(图7e,f)。总之,我们的数据表明升高葡萄糖水平触发小胶质细胞分泌损害DA神经元活力的因子,该表型在星形胶质细胞中也观察到。我们的结果指出胶质细胞在糖尿病神经系统并发症中的重要作用,并强调神经元-胶质细胞串扰作为未来治疗途径的干预靶点。
更多结果和补充图表:doi:10.1186/s13287-026-04965-y


扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:
https://cnsknowall.com
CNSknowall:24年最新问世的遥遥领先的科研数据(0代码生信+统计学)分析平台,同时含有机制图模块+汉化版Pubmed融合Deepseek高效筛选目标文献+SCI文献例句/语料检索模块+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动批阅!