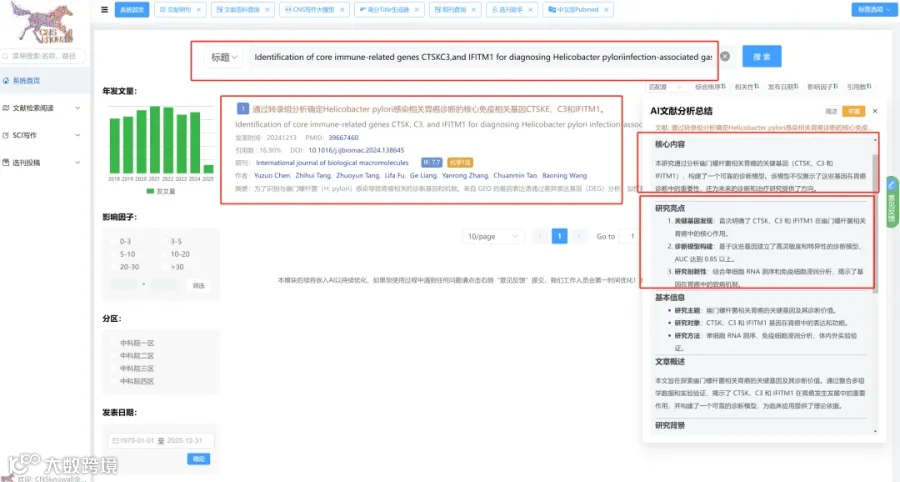
AD遗传风险因子SPI1(PU.1)高表达于小胶质细胞,但其细胞特异性功能一直成谜。本研究首次构建小胶质细胞条件性Spi1敲除的AD小鼠模型,发现Spi1缺失虽不改变APP加工,却通过严重削弱Aβ吞噬能力,导致淀粉样斑块沉积暴增、神经炎症加剧及轴突营养不良恶化。为破解机制,团队运用TMT标记定量蛋白组学,结合STRING互作网络分析,精准锁定“免疫反应-吞噬”通路异常,并创新性地通过功能回补实验证实,激活Syk、Lyn和Fcgr1三个靶基因可完全挽救Spi1敲低导致的Aβ吞噬缺陷。这颠覆了以往外周血研究结论,证明SPI1在脑内固有免疫中是保护性因子,其下游效应分子或成AD新药开发的黄金靶点。
今天给大家解读一篇4月发表在《Neuron》上的题目为“Deletion of SPI1 in microglia exacerbates amyloid pathology by impairing microglial response in Alzheimer's disease models.”的文章。该研究旨在阐明小胶质细胞特异性转录因子SPI1在阿尔茨海默病(AD)发病中的作用。研究人员利用条件性基因敲除技术,在表达淀粉样蛋白前体蛋白(APP)和早老素1(PS1)突变的小鼠模型(APPPS1-21)中,特异性地敲除了小胶质细胞中的Spi1基因。研究发现,Spi1的缺失显著增加了小鼠脑内的可溶性和不可溶性Aβ水平、淀粉样斑块沉积、胶质细胞增生以及营养不良性神经突的形成。机制上,Spi1缺失导致小胶质细胞对斑块的包裹减少,吞噬功能特别是对Aβ的摄取能力显著下降。通过蛋白质组学分析,研究鉴定出与吞噬作用相关的差异表达蛋白,并进一步通过功能实验证实,Spi1通过调控下游靶基因Syk、Lyn和Fcgr1的表达来影响小胶质细胞的吞噬功能。过表达这些基因能够有效挽救因Spi1缺失而受损的Aβ清除能力。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!包括数据集、绘图代码、图表复现、思路总结、参考文献……0代码!鼠标点点点即可轻松完成5-10分生信SCI全文复现!
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!



团队成员合影(位于上海陆家嘴中心,可随时预约参观)
题目:《小胶质细胞中SPI1的缺失通过损害小胶质细胞反应加剧阿尔茨海默病模型中的淀粉样蛋白病理》Deletion of SPI1 in microglia exacerbates amyloid pathology by impairing microglial response in Alzheimer's disease models
发表期刊:Neuron
影响因子:15
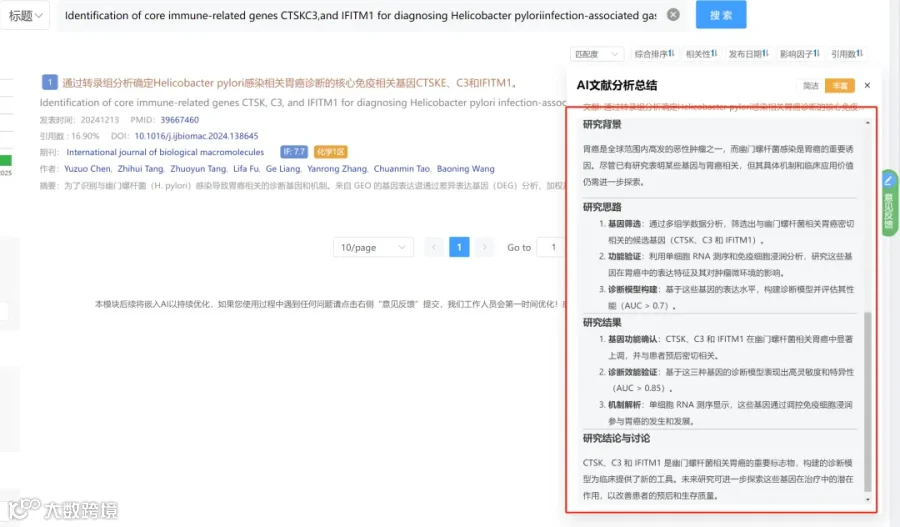
研究背景:
- AD与小胶质细胞
阿尔茨海默病(AD)的特征包括Aβ斑块、神经纤维缠结和神经营养不良。近期AD的全基因组关联研究(GWAS)发现了多个免疫相关的风险基因,其中小胶质细胞作为中枢神经系统的专业吞噬细胞,在AD发病中扮演重要角色。
- SPI1的潜在角色
转录因子PU.1(由
SPI1基因编码)主要由中枢神经系统中的小胶质细胞表达,已被报道为AD的遗传风险因素。然而,小胶质细胞中SPI1在AD发病中的具体作用尚不清楚。
- 知识缺口
先前的研究使用了全身性
Spi1敲低或过表达模型,虽然提供了宝贵见解,但无法区分Spi1在不同细胞类型(特别是小胶质细胞和外周免疫细胞)中的特异性作用。本研究旨在填补这一知识空白。
CNSknowall 平台 Pubmed+AI 快速提炼全文要点
研究思路:
- 构建细胞特异性模型
将
Spi1条件性敲除小鼠(Spi1fl/fl)与在诱导剂他莫昔芬作用下可特异性敲除小胶质细胞基因的Cx3cr1-CreERT2小鼠交配,获得小胶质细胞特异性Spi1敲除小鼠(Spi1ΔMG)。
- 评估对AD病理的影响
将
Spi1ΔMG小鼠与AD模型小鼠(APPPS1-21)交配,系统评估Spi1缺失对Aβ水平、淀粉样斑块沉积、胶质细胞增生和神经营养不良等病理特征的影响。
- 探究细胞机制
通过体内(MeX04注射)和体外(Aβ-pHrodo摄取实验)的吞噬功能分析,考察
Spi1缺失对小胶质细胞吞噬Aβ能力的影响。
- 寻找分子机制
对
Spi1敲除小鼠的大脑皮层进行深度蛋白质组学分析,鉴定差异表达蛋白(DEPs)和关键信号通路。
- 验证关键靶点
基于蛋白质组学和ChIP-Seq数据,筛选出
Spi1调控的、与吞噬作用相关的候选基因。通过体外过表达实验,验证哪些候选基因的恢复能够挽救Spi1敲低导致的Aβ吞噬障碍。
研究亮点:
- 细胞特异性研究
利用条件性敲除(
Cx3cr1-CreERT2)小鼠模型,首次在体内明确了小胶质细胞中Spi1缺失对AD病理加剧的直接作用,避免了全基因组敲除模型中其他细胞类型的影响。
- 机制解析
通过蛋白质组学与功能验证相结合的方法,不仅发现了
Spi1缺失导致的蛋白质组变化,还具体鉴定并验证了Syk、Lyn和Fcgr1作为其下游关键效应分子,它们共同调控小胶质细胞的吞噬功能。
- 功能挽救
研究不仅证明了问题,还提供了解决方案。通过在体外实验中过表达
Syk、Lyn和Fcgr1,成功恢复了因Spi1敲低而受损的Aβ摄取能力,为AD治疗提供了潜在的新靶点。
研究结果:
- 模型验证
Spi1ΔMG小鼠大脑中Spi1的mRNA和蛋白水平显著降低,小胶质细胞的突起数量减少,但小胶质细胞的总数未受影响。
- 加重Aβ病理
在APPPS1-21背景下,
Spi1缺失导致大脑皮层和海马中不可溶及可溶性Aβ40和Aβ42水平显著升高。
- 增加淀粉样斑块沉积
Spi1缺失显著增加了大脑皮层和海马中总Aβ斑块(82E1+)和纤维状斑块(X-34+)的负荷和数量。
- 加剧胶质增生
Spi1缺失导致小胶质细胞(IBA1+)和星形胶质细胞(GFAP+)的激活区域和数量显著增加,并伴随促炎细胞因子(Il1b、iNos)mRNA水平的升高。
- 损害小胶质细胞反应和吞噬功能
-
在体内,
Spi1缺失导致围绕纤维斑块的小胶质细胞簇面积减少。
-
在体内Aβ吞噬实验中,
Spi1缺失的小胶质细胞摄取MeX04标记的Aβ能力显著降低。
-
在体外,
Spi1敲低的原代小胶质细胞对fAβ-pHrodo的摄取能力显著下降,且这种下降并非由细胞活力降低引起。
- 蛋白质组学分析
Spi1缺失导致199个DEPs,其中与“免疫反应_吞噬作用”相关的网络是最显著富集的通路。
- 下游靶点验证
-
在BV-2细胞中,
Spi1敲低降低了Syk、Lyn、Stat1、Entpd1和Fcgr1的mRNA水平。
- 过表达
Syk、Lyn或Fcgr1均能显著恢复因Spi1敲低而受损的Aβ摄取能力。
-
在原代小胶质细胞中同样验证了
Spi1敲低会降低Syk、Lyn和Fcgr1的表达,且过表达Syk和Fcgr1能挽救Aβ吞噬功能。
研究总结:
- 核心结论
小胶质细胞中转录因子SPI1对于维持其在AD病理中的正常功能至关重要。
Spi1缺失通过下调其下游靶基因Syk、Lyn和Fcgr1,损害小胶质细胞的吞噬功能,特别是对Aβ的清除能力,从而加剧了AD相关的淀粉样蛋白病理、胶质增生和神经元损伤。
- SPI1的“双重作用”
研究指出,SPI1在AD中扮演着双刃剑的角色。一方面,敲低SPI1可减弱体外炎症反应;但另一方面,本研究体内实验表明,SPI1缺失会因显著降低Aβ清除能力而加剧病理。因此,未来的治疗策略需要谨慎平衡SPI1的抗炎和促吞噬功能。
- 与人类遗传学研究的关联与矛盾
本研究结果似乎与一项人类遗传学研究相悖,该研究报道了与较低SPI1表达相关的保护性变异。作者解释,这可能是由于SPI1在外周血单核细胞和大脑小胶质细胞中的表达相关性较低所致。本研究直接证明了在脑内小胶质细胞中降低SPI1表达会恶化AD病理。
- 潜在治疗靶点
本研究揭示的
SPI1-Syk/Lyn/Fcgr1信号轴,特别是恢复这些下游靶基因的功能,可能成为AD治疗的一个有前景的新策略。同时,研究中发现的APOE水平升高也提示了SPI1-APOE调控轴的重要性。
- 研究局限性
研究指出了
Cx3cr1CreER小鼠可能存在的“泄露”活性问题,但作者通过实验设计(向所有实验组注射他莫昔芬)和对照实验(确认4-OHT对Spi1野生型无影响)对此进行了控制。此外,研究未能排除在长寿命血管周巨噬细胞中发生脱靶效应的可能性。
结果译文:
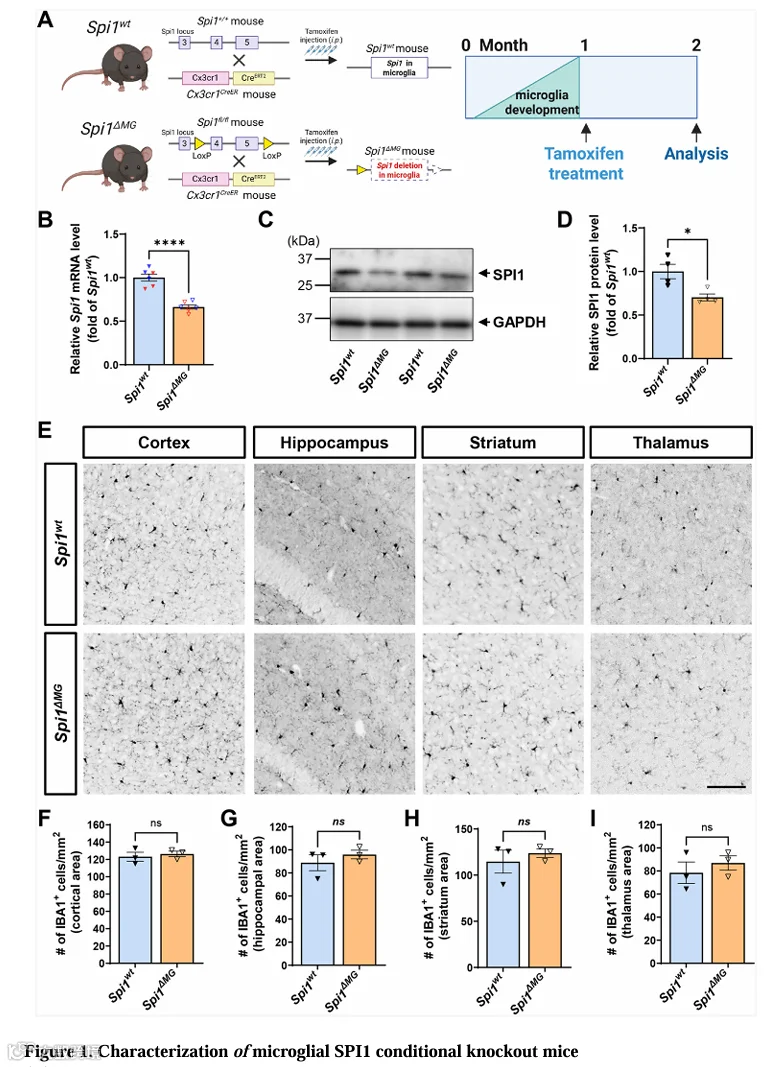
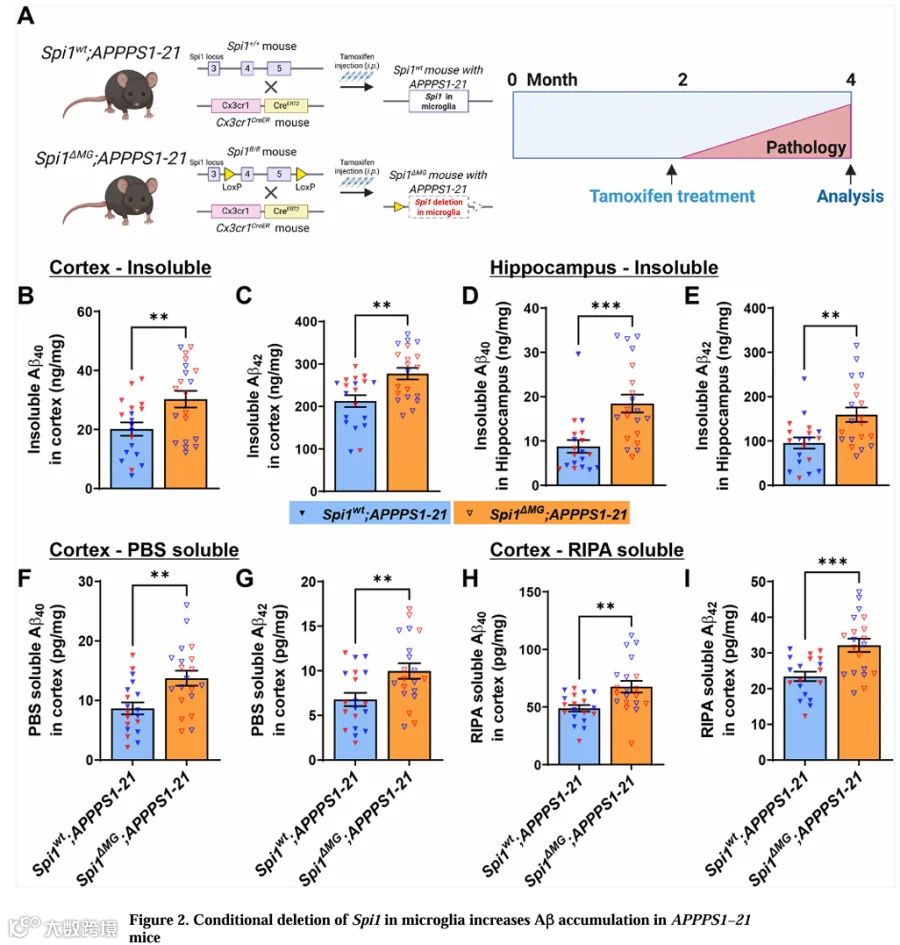
在评估小胶质细胞Spi1缺失对AD病理特征的影响前,我们验证了小胶质细胞类型特异性Spi1条件性敲除小鼠模型。我们对4周龄的Spi1 MG和Spi1wt小鼠连续5天给予他莫昔芬,随后在小鼠2月龄时处死(图1A)。为验证Spi1缺失,我们进行了qPCR和Western blot分析以测定脑内Spi1水平。qPCR结果显示Spi1 MG小鼠脑内Spi1 mRNA水平比Spi1wt小鼠低33.7%(图1B)。Western blot分析测定SPI1蛋白水平在Spi1 MG小鼠中较Spi1wt小鼠低29.8%(图1C和1D)。为进一步确定Spi1缺失是否有效敲除了小胶质细胞中的Spi1,我们用抗SPI1和抗IBA1抗体进行了脑切片染色(图S1A)。与Spi1wt小鼠相比,Spi1 MG小鼠皮层区域SPI1阳性小胶质细胞数量显著减少42.7%(图S1B)。此外,骨架分析揭示了Spi1 MG小鼠脑内小胶质细胞突起数量显著减少(图S1C和S1D)。Spi1表达的这种降低证实了我们条件性敲除策略的有效性。
由于PU.1对胚胎期小胶质细胞前体阶段的小胶质细胞发育至关重要,在该早期阶段删除Spi1基因会干扰小胶质细胞成熟。然而,由于本研究中Spi1缺失是在小胶质细胞成熟后诱导的,我们假设小胶质细胞密度在Spi1 MG与Spi1wt小鼠间无差异。我们检测了成熟后Spi1缺失是否对小胶质细胞群体密度有意外影响。用抗IBA1抗体进行脑切片染色(图1E和S1E)。最终他莫昔芬注射后4周,多个脑区的IBA1⁺细胞数量未因小胶质细胞Spi1基因丢失而改变(图1F-1I)。此外,最终他莫昔芬注射后1或2周,Spi1缺失也未影响IBA1⁺细胞负荷或数量(图S1F和S1G)。这证明尽管脑内Spi1水平降低,但Spi1 MG小鼠小胶质细胞数量未受影响。
综上,这些发现表明虽然小胶质细胞SPI1缺乏不改变小胶质细胞数量,但导致小胶质细胞突起减少。这提示小胶质细胞监视活动普遍下降或其功能潜在改变。
2.小胶质细胞Spi1缺失增加APPPs1-21小鼠Aβ水平
为研究SPI1在AD病理特征中的细胞类型特异性作用,我们构建了携带APPPS1-21转基因的小胶质细胞特异性Spi1敲除小鼠(Spi1 MG;APPPS1-21)及对照小鼠(Spi1wt;APPPS1-21)。APPPS1-21小鼠模型约在6-8周龄时开始在新皮层积累Aβ,并在4月龄时表现出稳固的Aβ积累和斑块沉积。因此,我们在小鼠7周龄时连续5天给予他莫昔芬,并在4月龄时进行评估(图2A)。
为确定小胶质细胞Spi1缺失对AD病理特征的影响,我们用Aβ电化学发光法测定了Spi1wt;APPPS1-21和Spi1 MG;APPPS1-21小鼠皮层和海马组织中不可溶性Aβ40和Aβ42的水平(图2B-2E)。与Spi1wt;APPPS1-21小鼠相比,小胶质细胞中Spi1敲低分别使皮层不可溶性Aβ40和Aβ42水平显著增加了1.48倍和1.31倍(图2B和2C)。同样,海马中Aβ40和Aβ42水平分别显著增加2.11倍和1.67倍(图2D和2E)。此外,我们分别分析了雄性和雌性数据,发现小胶质细胞Spi1缺失在相似程度上增加了雄性和雌性小鼠皮层和海马中的不可溶性Aβ水平(图S2A-S2D)。
除不可溶性Aβ外,我们定量了这些小鼠皮层组织PBS和RIPA组分中的可溶性Aβ40和Aβ42水平。在PBS可溶性组分中,Spi1 MG;APPPS1-21小鼠Aβ40水平增加1.58倍(图2F),Aβ42水平增加1.47倍(图2G)。在RIPA可溶性组分中,Aβ40水平增加1.38倍(图2H),Aβ42水平增加1.37倍(图2I)。此外,这些小鼠皮层和海马组织中的不可溶性Aβ40和Aβ42水平之间存在显著相关性(图S2E和S2F),皮层组织PBS和RIPA组分中的亦如此(图S2G和S2H)。综上,我们的数据证明小胶质细胞Spi1缺失导致多个脑组分中Aβ水平增加。
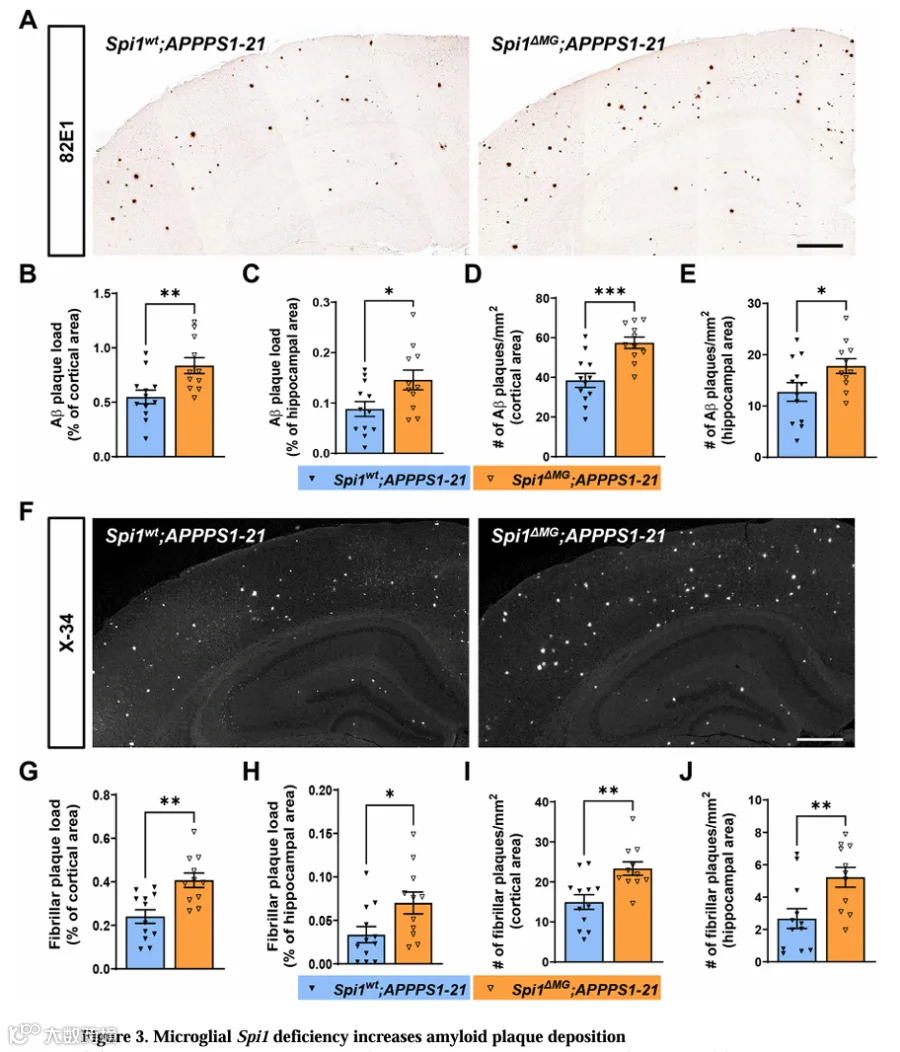
我们继而评估了小胶质细胞Spi1缺失是否会改变淀粉样斑块沉积程度。我们用82E1抗体染色脑切片。与生化Aβ测定结果一致(图2B-2E),小胶质细胞Spi1缺失使皮层和海马区域的Aβ斑块负荷分别显著增加1.52倍和1.65倍(图3B和3C)。此外,在Spi1 MG;APPPS1-21小鼠皮层和海马中,经面积标准化的Aβ斑块数量也显著增加(图3D和3E)。
我们进一步用X-34染色脑切片评估纤维性淀粉样斑块负荷(图3F)。观察到Spi1 MG;APPPS1-21小鼠皮层和海马区域纤维性斑块负荷分别显著增加1.69倍和2.08倍(图3G和3H)。经面积标准化的纤维性斑块数量也显著增加(图3I和3J)。综上,这些数据证明小胶质细胞Spi1缺失显著增加APPPS1-21小鼠脑内淀粉样斑块沉积。
4.小胶质细胞Spi1缺失不改变参与APP蛋白水解加工的蛋白水平
我们发现小胶质细胞中Spi1缺失增加Aβ水平和淀粉样斑块沉积。这些增加可能是由参与APP加工的蛋白水平变化引起的。为检测小胶质细胞Spi1缺失是否影响APP蛋白的水解加工,我们通过Western blot分析测定了全长APP、其切割酶BACE1和APP-C末端片段(β-CTF)的蛋白水平(图S2I)。未观察到APP、BACE1和β-CTF蛋白水平在Spi1 MG;APPPS1-21小鼠皮层中有显著差异(图S2J-S2L)。这些数据表明小胶质细胞中Spi1缺失不影响APP加工。
5.小胶质细胞中Spi1缺失增加小胶质细胞增生和星形胶质细胞增生
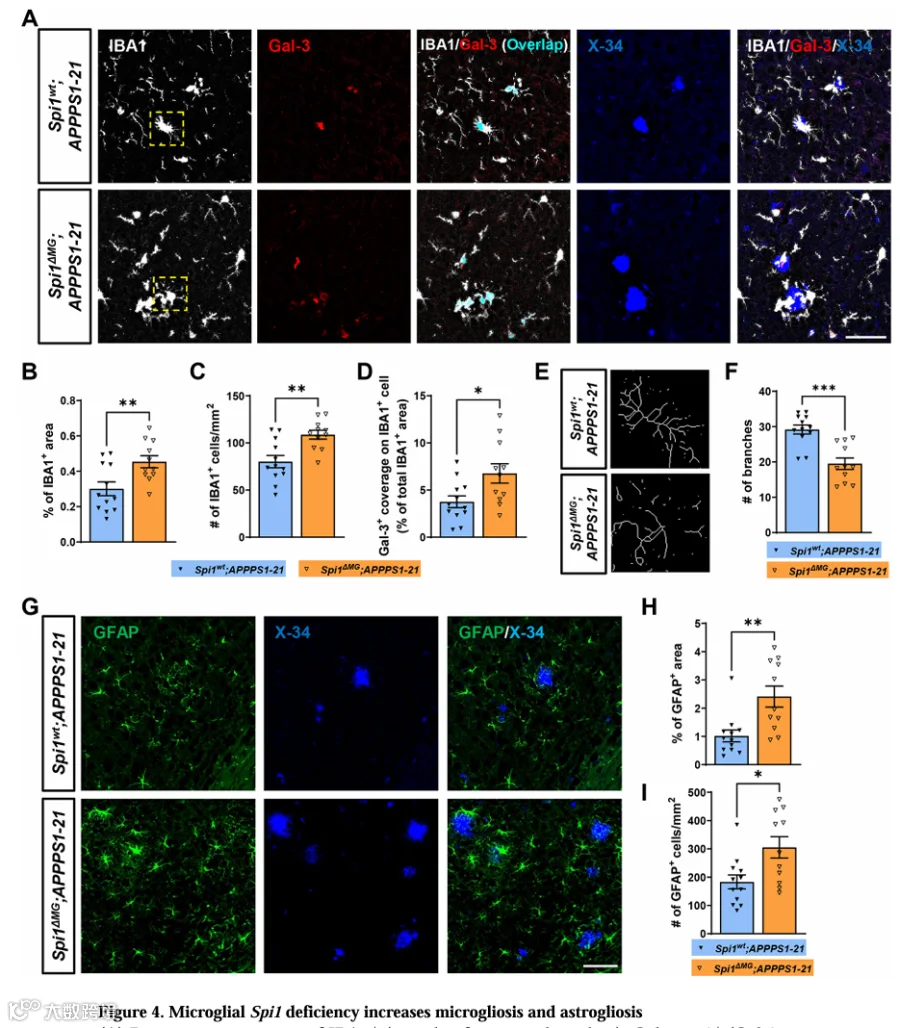
小胶质细胞和星形胶质细胞的异常激活在AD患者和APPPS1-21转基因模型小鼠脑内被观察到。因此,我们评估了小胶质细胞Spi1丢失是否影响胶质细胞激活。Galectin-3(Gal-3)是参与神经炎症的关键蛋白,在神经退行性条件下强烈上调。我们用抗IBA1和抗Gal-3抗体染色小鼠脑切片以评估斑块周围小胶质细胞激活,并用X-34染色以识别纤维性斑块(图4A)。与Spi1wt;APPPS1-21小鼠相比,Spi1 MG;APPPS1-21小鼠皮层区域IBA1⁺细胞覆盖面积和IBA1⁺每面积细胞数分别显著增加1.51倍和1.36倍(图4B和4C)。
此外,与Spi1wt;APPPS1-21小鼠相比,Spi1 MG;APPPS1-21小鼠皮层IBA1⁺细胞区域内Gal-3⁺细胞占据面积显著增加1.80倍(图4D)。小胶质细胞形态学骨架分析揭示Spi1 MG;APPPS1-21小鼠每小胶质细胞突起数少于Spi1wt;APPPS1-21小鼠(图4E和4F)。对于星形胶质细胞增生,通过GFAP染色测定(图4F),与Spi1wt;APPPS1-21小鼠相比,Spi1 MG;APPPS1-21小鼠皮层GFAP⁺细胞面积和经面积标准化的GFAP⁺细胞数量分别显著增加2.39倍(图4G)和1.57倍(图4H)。
此外,为探究小胶质细胞Spi1丢失是否影响神经炎症,我们使用qPCR测定了促炎细胞因子和其他神经炎症标志物的水平。结果表明Il1b和iNos表达水平在Spi1 MG;APPPS1-21小鼠中显著增加(图S3A和S3B),而Tnf显示增加趋势但未达到统计显著性(p=0.0534,图S3C)。此外,Gfap mRNA表达在Spi1 MG;APPPS1-21小鼠中显著增加(图S3D),与GFAP组织学分析一致(图4G-4I)。
综上,我们的结果表明小胶质细胞中Spi1缺失减少了Aβ淀粉样小鼠模型中其突起,损害其检测和响应脑微环境变化或损伤的能力。这种损害可能因此导致小胶质细胞和星形胶质细胞激活增加,在Aβ淀粉样变性小鼠模型中触发神经炎症。
6.小胶质细胞中SPI1缺失减少斑块周围小胶质细胞并降低吞噬活性
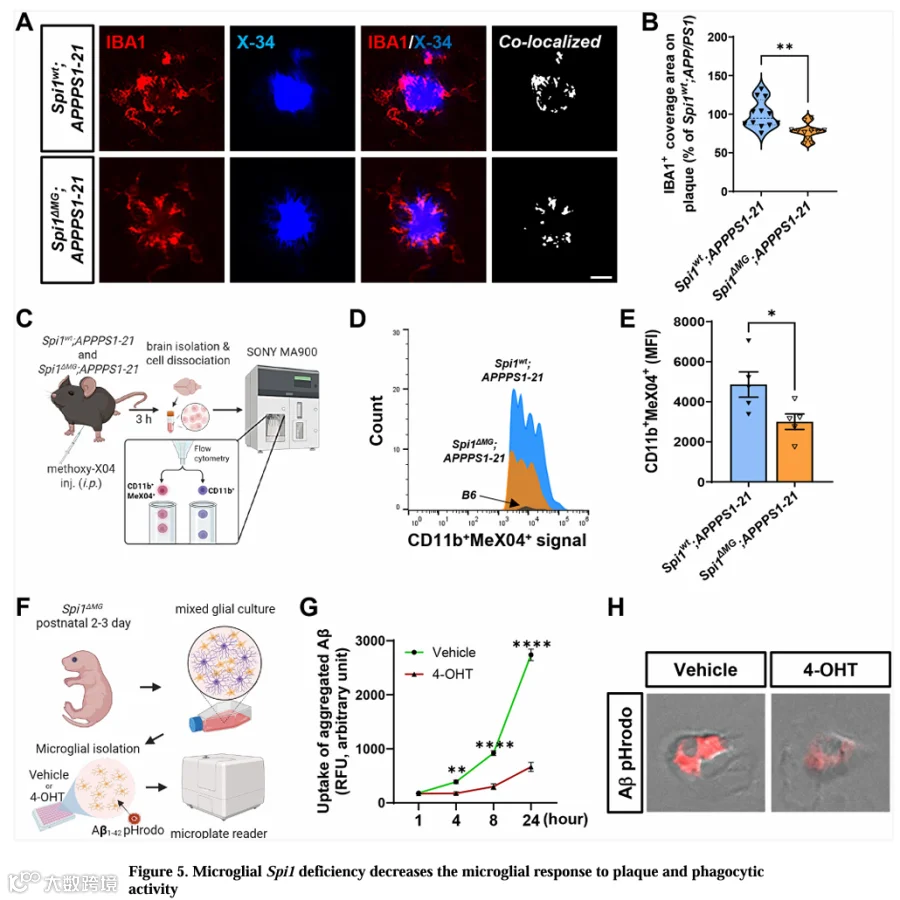
激活的小胶质细胞环绕淀粉样斑块,参与通过吞噬作用清除Aβ。为探究小胶质细胞Spi1缺失是否影响此过程,我们分析了斑块相关小胶质细胞。用IBA1抗体和X-34淀粉样斑块染料共染脑切片(图5A),并定量IBA1⁺细胞与每个斑块共定位的面积。与Spi1wt;APPPS1-21小鼠相比,Spi1 MG;APPPS1-21小鼠IBA1⁺小胶质细胞簇覆盖的斑块面积显著减少22.7%(图5B)。
此外,为确定Spi1缺失是否改变小胶质细胞吞噬能力,我们进行了体内Aβ吞噬测定。对小鼠腹腔注射methoxy-X04(MeX04),3小时后获取大脑并分离小胶质细胞,通过流式细胞术分析MeX04⁺ Aβ被小胶质细胞摄取的程度(图5C和图S4A-S4D)。尽管Spi1 MG;APPPS1-21小鼠fAβ负荷更高(图3F-3J),但小胶质细胞Spi1缺失显著降低摄取了Aβ的CD11b⁺MeX04⁺小胶质细胞的中位荧光强度(MFI)(图5D和5E)。我们还观察到Spi1 MG;APPPS1-21小鼠中CD11b⁺MeX04⁺小胶质细胞的比例呈下降趋势(图S4E)。这些发现提示小胶质细胞中SPI1缺失可能损害fAβ吞噬作用。
为加强我们的体内Aβ吞噬测定结果(图5D和5E),我们还用fAβ进行了体外Aβ吞噬测定。从Spi1 MG小鼠分离原代小胶质细胞,用载体或1μM 4-OHT预处理4天,然后与pHrodo Red染料偶联的100nM fAβ孵育24小时。我们首先验证了4-OHT介导的Spi1缺失的效果。与载体处理的原代小胶质细胞相比,4-OHT处理组Spi1 mRNA水平降低79.7%(图S5A)。接下来,我们纵向评估了Spi1基因敲低后原代小胶质细胞对fAβ-pHrodo的摄取(图5F)。与载体组相比,Spi1敲低(4-OHT)组在fAβ-pHrodo处理后4、8和24小时,fAβ摄取分别显著降低54.3%、67.3%和75.7%(图5G)。我们还目视确认了在fAβ-pHrodo处理后8小时,4-OHT组fAβ摄取减少(图5H)。为排除fAβ摄取降低仅因Spi1敲低后细胞活力降低,我们评估了细胞活力,未观察到显著差异(图S5C)。我们还评估了酵母聚糖颗粒的摄取,Spi1敲低使其显著减少33.1%(图S5D),且无细胞活力差异(图S5E)。这些发现提示Spi1缺失损害一般小胶质细胞吞噬功能而不影响细胞活力。
7.小胶质细胞Spi1丢失加剧轴突营养不良并增加神经炎性斑块
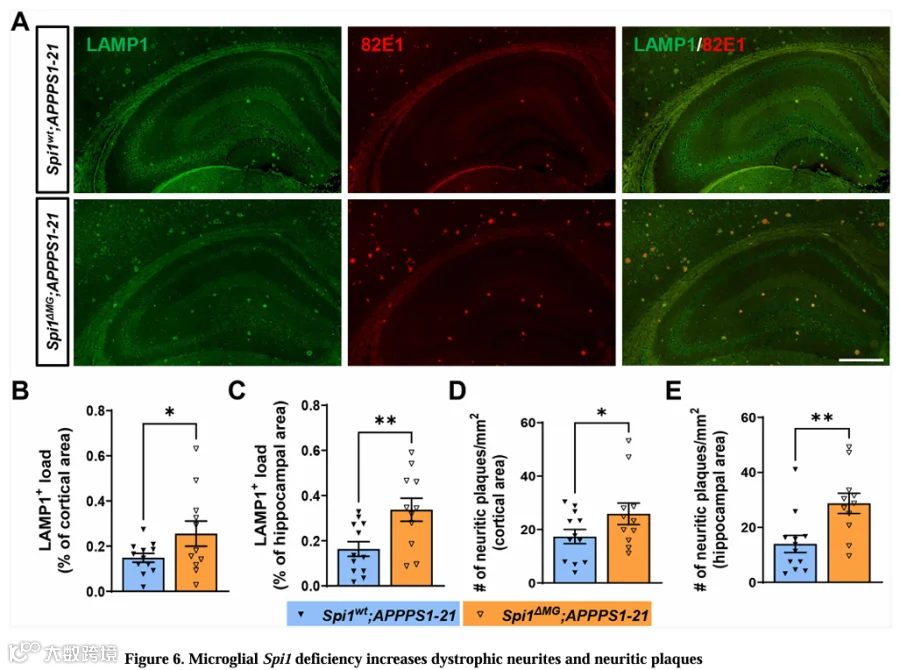
营养不良性神经突起是AD患者脑内显著的神经病理特征。这些是围绕淀粉样斑块的异常神经突起。因此,评估其程度作为斑块相关神经元毒性的重要读数。最近研究表明,围绕淀粉样斑块的神经突起富含溶酶体标志物LAMP1。我们用抗LAMP1抗体评估小胶质细胞Spi1缺失是否影响营养不良性神经突起沉积,并用82E1抗体共染评估神经炎性斑块沉积(图6A)。小胶质细胞Spi1缺失显著使皮层和海马区域LAMP1⁺簇负荷分别增加1.72倍和2.06倍(图6B和6C)。此外,LAMP1⁺簇主要围绕82E1⁺ Aβ斑块被观察到(图6A)。Spi1 MG;APPPS1-21小鼠皮层和海马区域神经炎性斑块(LAMP1⁺82E1⁺)数量也分别显著增加1.45倍和2.05倍(图6D和6E)。这些结果表明小胶质细胞Spi1缺失显著增加营养不良性神经突起范围和神经炎性斑块数量。
8.蛋白组分析提示小胶质细胞Spi1缺失通过损害吞噬功能加剧淀粉样病变
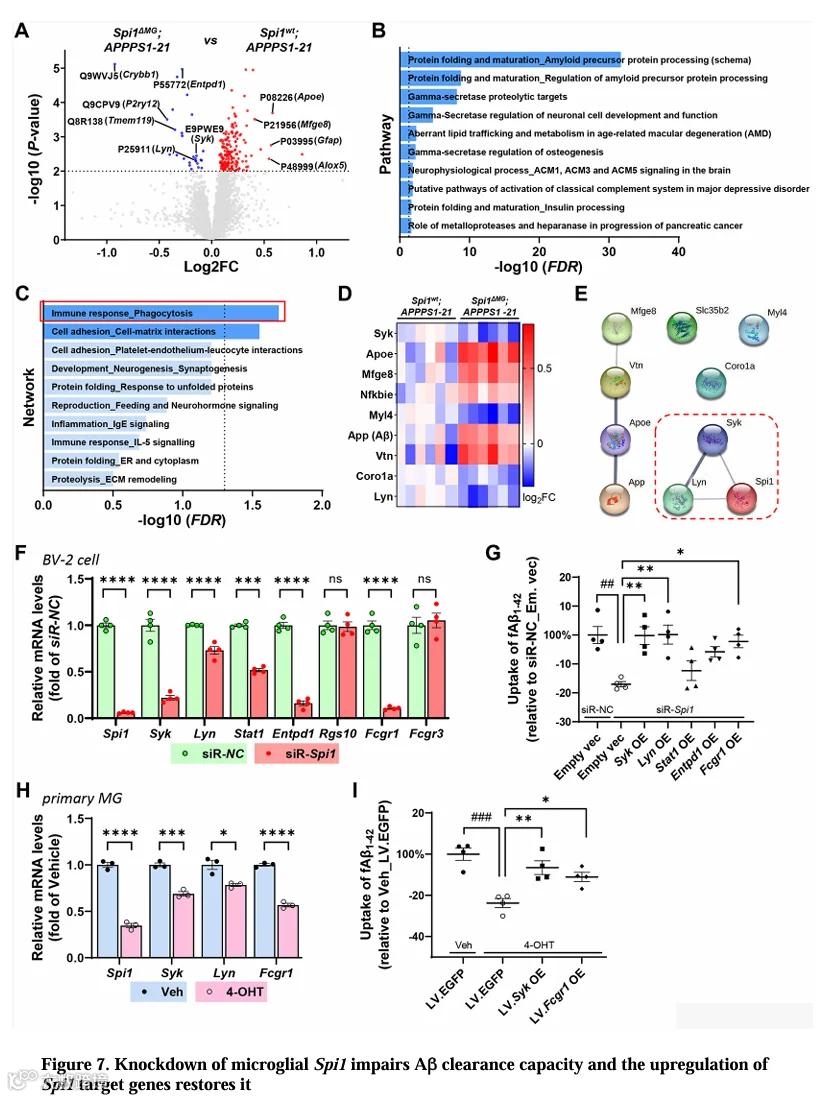
由于小胶质细胞Spi1缺失不影响APP加工,我们接下来探索了导致淀粉样病变表型的其他机制。为确定Spi1缺失引起的蛋白组变化,我们使用TMT标记质谱对Spi1wt;APPPS1-21和Spi1 MG;APPPS1-21小鼠皮层进行了深度蛋白组分析。我们定量了9,592个独特肽段(9,084个基因)和199个差异表达蛋白(DEPs)(p < 0.01,图7A中水平虚线所示)(表S1)。在这些DEPs中,我们观察到小胶质细胞Spi1缺失后小胶质细胞稳态蛋白下调,而APOE(一种疾病相关小胶质细胞标志物)显著上调。我们还观察到MFGE8、ALOX5和GFAP表达显著增加。星形胶质细胞标志物GFAP的显著上调与GFAP组织学分析一致。
为理解Spi1 MG;APPPS1-21小鼠中上调和下调DEPs所调控的特定生物学通路,我们使用MetaCore™生物信息学方法进行了通路和网络富集分析。“蛋白质折叠和成熟_淀粉样前体蛋白加工”通路(图7B和表S2)和“免疫反应_吞噬作用”网络(图7C和表S3)被识别为最显著改变的通路和网络。通路术语“蛋白质折叠和成熟_淀粉样前体蛋白加工”包含Aβ40、Aβ42作为组分,进一步支持我们的观察。我们深入研究了“免疫反应_吞噬作用”网络的分子基础(图7C)。在该网络组分中,我们在蛋白组数据集中识别出11个网络对象中的9个DEP(图7D和表S4)。为确定这些DEP与Spi1之间潜在的功能和物理蛋白关联,我们使用STRING数据库进行互作分析(图7E)。该分析进一步支持SPI1在功能或物理上与“免疫反应_吞噬作用”网络中的Syk和Lyn相关联。这一整合系统生物学方法证明小胶质细胞Spi1缺失通过干扰吞噬功能影响AD病理。
9.Spi1功能丢失通过Syk、Lyn和Fcgr1损害Aβ摄取
我们进一步研究了激活Spi1的靶基因是否能恢复因小胶质细胞Spi1敲除受损的Aβ摄取(图5D,5E,5G,5H)。基于“免疫反应_吞噬作用”组分和Spi1 ChIP-Seq数据,我们筛选了七个由Spi1直接靶向且可能参与吞噬作用的基因(Syk, Lyn, Stat1, Entpd1, Rgs10, Fcgr1和Fcgr3)。我们首先检查了我们的蛋白组数据集中这些基因的蛋白水平,发现它们在Spi1 MG;APPPS1-21小鼠中显著降低(图S7A)。接下来,我们在BV-2小胶质细胞中使用siRNA敲低Spi1后测定这些基因的表达水平。其中,Syk、Lyn、Stat1、Entpd1和Fcgr1的表达水平因Spi1敲低而显著降低,而Rgs10和Fcgr3未改变(图7F)。
基于这些筛选数据,我们进一步评估了过表达Syk、Lyn、Stat1、Entpd1和Fcgr1能否恢复因Spi1敲低受损的Aβ摄取。我们在BV-2小胶质细胞中Spi1敲低后过表达这些候选基因,并评估fAβ-pHrodo摄取水平。与siRNA阴性对照组相比,Spi1敲低组fAβ摄取显著降低(图7G)。在候选基因中,Syk、Lyn和Fcgr1的过表达显著恢复了BV-2小胶质细胞中因Spi1敲低受损的Aβ摄取(图7G)。
为在Spi1 MG小鼠原代小胶质细胞中验证我们从BV-2小胶质细胞系得到的发现,我们用1μM 4-OHT诱导Spi1敲低后测定了Syk、Lyn和Fcgr1的表达水平。与我们在BV-2细胞中的发现一致,Spi1敲低显著降低了原代小胶质细胞中Syk、Lyn和Fcgr1的表达(图7H)。接下来,我们旨在确定在原代小胶质细胞中过表达Syk、Lyn和Fcgr1能否恢复Spi1敲低条件下的Aβ摄取。由于技术限制,我们无法在原代小胶质细胞中实现Lyn过表达。因此,我们聚焦于评估Spi1敲低条件下过表达Syk和Fcgr1能否挽救fAβ摄取。对原代小胶质细胞fAβ-pHrodo摄取的分析显示,与载体组相比,Spi1敲低(4-OHT)组fAβ摄取显著降低(图7I)。重要的是,与我们在BV-2细胞中的发现一致,Syk和Fcgr1的过表达在原代小胶质细胞中显著恢复了因Spi1敲低受损的Aβ摄取(图7I),且不影响细胞活力(图S7C)。综上,这些数据证明激活某些Spi1靶基因可恢复因Spi1敲低受损的小胶质细胞吞噬功能。
更多结果和补充图表:doi: 10.1016/j.neuron.2026.03.011
长按二维码关注我们,用最短的时间和最高的效率学习更多数据分析方法!
扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:
https://cnsknowall.com
CNSknowall:24年最新问世的遥遥领先的科研数据(0代码生信+统计学)分析平台,同时含有机制图模块+汉化版Pubmed融合Deepseek高效筛选目标文献+SCI文献例句/语料检索模块+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动批阅!