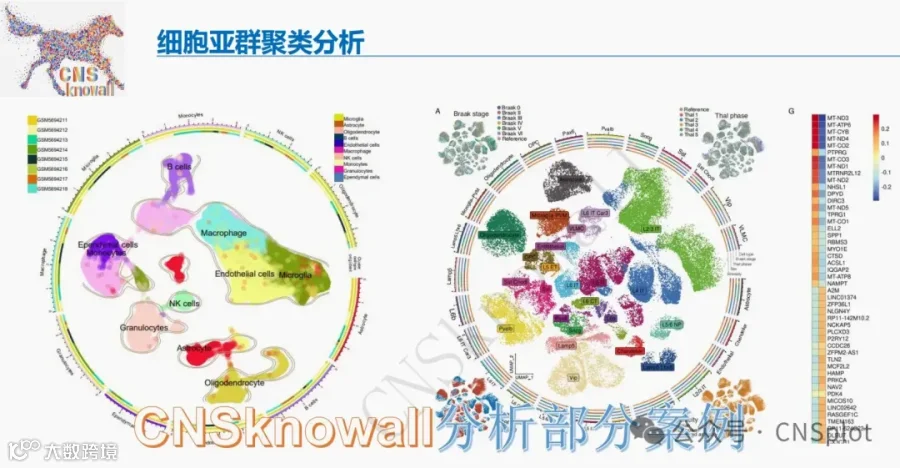
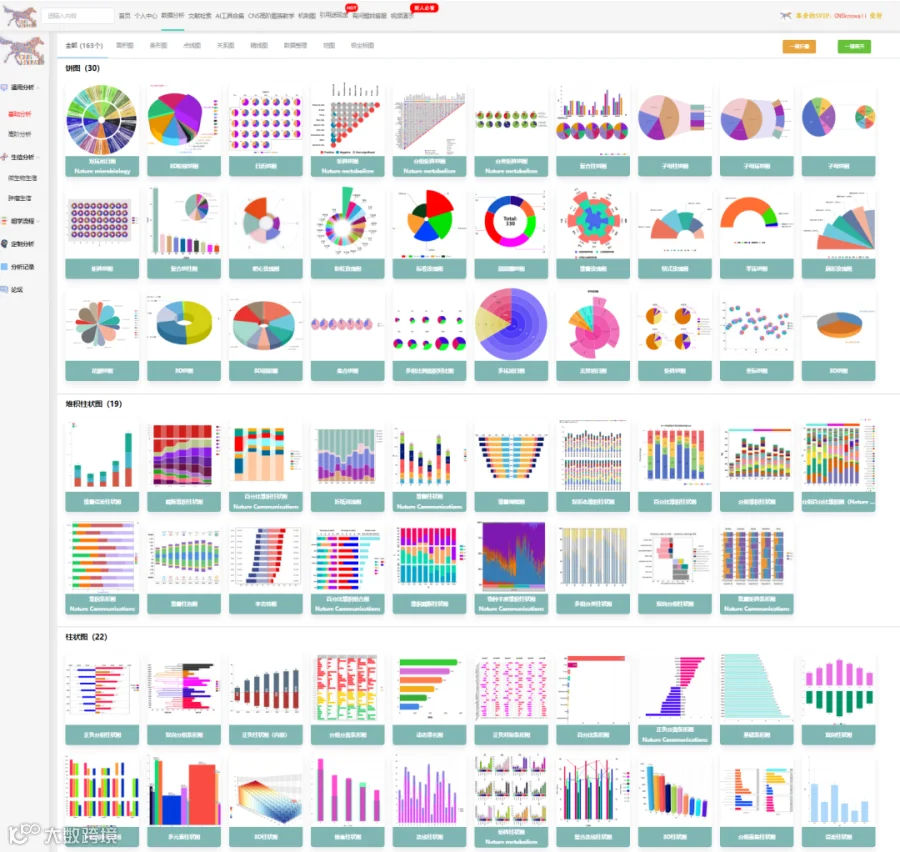
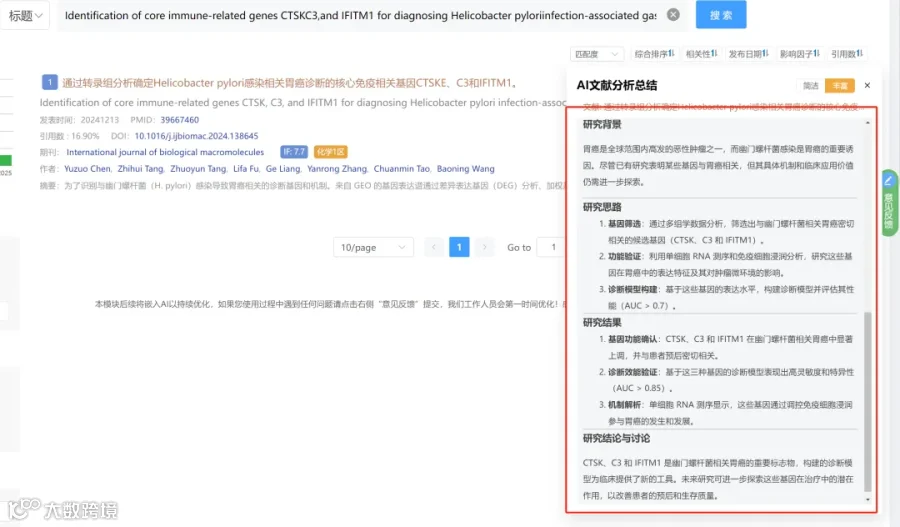
今天给大家解读一篇2月发表在《Cells》上的题目为“Phosphatidylcholine and CHPT1 as Central Drivers of Chemoresistance in Colorectal Cancer: Lipidomic and Functional Insights.”的文章。本研究旨在阐明磷脂酰胆碱代谢及其生物合成调节因子CHPT1在结直肠癌化疗耐药中的作用。通过对比敏感与耐药细胞系,研究者发现耐药细胞具有更高的PC含量和改变的PC/LPC比例,并证明CHPT1过表达足以导致PC积累和耐药表型。临床数据分析显示CHPT1高表达与患者不良预后相关。机制上,CHPT1驱动的PC富集可能维持促生存信号。最后,研究证明药物edelfosine可通过干扰Kennedy通路,增强耐药模型对化疗的敏感性。文章结论认为,CHPT1和PC代谢是结直肠癌药物反应的核心决定因素,靶向该通路是克服耐药的有前景的策略。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!
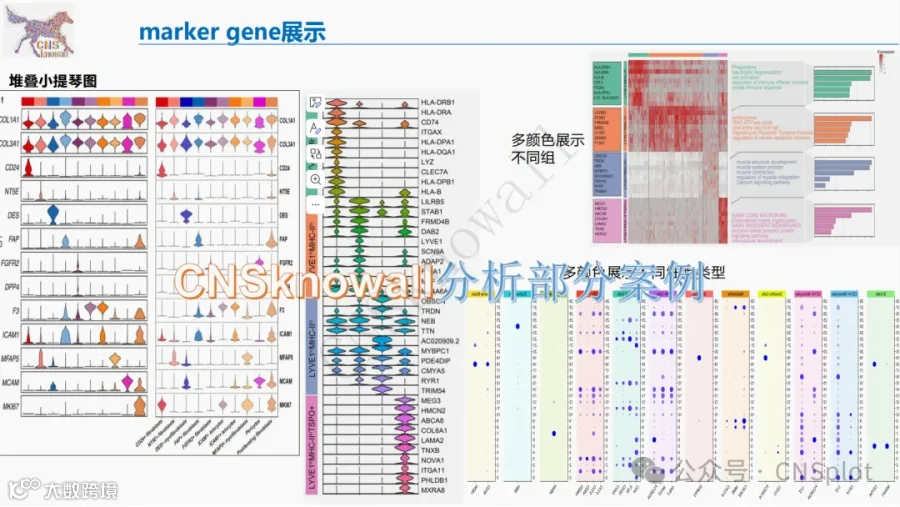
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!









团队成员合影(位于上海陆家嘴中心,可随时预约参观)





题目:《磷脂酰胆碱和CHPT1作为结直肠癌化疗耐药性的核心驱动因素:脂质组学和功能见解》Phosphatidylcholine and CHPT1 as Central Drivers of Chemoresistance in Colorectal Cancer: Lipidomic and Functional Insights
发表期刊:Cells
影响因子:5.2
研究背景:
脂质代谢重编程是恶性肿瘤的标志,支持肿瘤生长、生存和耐药。胆碱代谢是癌症生物学中的关键途径,PC是细胞膜中最丰富的磷脂。PC生物合成主要有两条途径:Lands循环和Kennedy通路。先前研究表明,LPCAT2通过促进脂滴形成在结直肠癌化疗耐药中起关键作用。Kennedy通路是PC从头合成的主要途径,CHPT1催化其最后一步反应。证据表明,CHPT1在其他癌症中是治疗抵抗的潜在驱动因子,但其在结直肠癌中的具体作用尚不明确。
研究思路:
- 模型选择
选用已被广泛证实具有稳定耐药差异的结直肠癌细胞对(耐药HT29 vs. 敏感SW620)作为研究系统。 - 表征分析
通过脂质组学(LC-MS/MS、GC-MS)比较两细胞系的磷脂组成;通过qPCR、Western Blot和酶活实验分析PC代谢相关酶的表达与活性。 - 功能验证
- 脂质功能
外源性补充不同磷脂,验证PC在诱导耐药中的特异性作用。 - 基因功能
在敏感SW620细胞中稳定过表达CHPT1,评估其对PC代谢、化疗敏感性(IC50测定)、DNA损伤/凋亡信号和内质网应激反应的影响。 - 临床相关性
利用人类蛋白质图谱数据库,分析CHPT1在结直肠癌组织中的表达及其与患者生存的关系;利用NCI-60数据集分析CHPT1表达与药物敏感性的相关性。 - 治疗探索
在耐药HT29细胞中,评估靶向PC代谢的药物edelfosine的细胞毒性、对其上游酶表达的影响,及其与标准化疗方案(FOX)的协同效应(使用Chou-Talalay法计算联合指数)。
研究亮点:
- 关键发现
CHPT1驱动的磷脂酰胆碱重塑是结直肠癌化疗耐药的关键代谢决定因素。 - 临床关联
CHPT1过表达促进多药耐药,并与结直肠癌患者不良生存相关。 - 代谢特征
脂质组学分析鉴定出改变的PC和LPC水平是耐药细胞的代谢特征。 - 治疗策略
Edelfosine通过靶向Kennedy通路,使CHPT1高表达的耐药结直肠癌细胞对化疗重新敏感。
研究结果:
- 脂质与酶学特征
耐药HT29细胞比敏感SW620细胞具有显著更高的PC、LPC等磷脂含量,但二酰甘油水平降低。HT29细胞中CHPT1在mRNA和蛋白水平均显著高表达,且磷脂酶C活性降低。 - PC的耐药作用
外源性补充PC(而非其他磷脂或溶血磷脂)能特异性保护敏感SW620细胞免受5-FU杀伤,证明PC积累是耐药的关键效应因子。 - CHPT1的核心功能
-
在SW620细胞中过表达CHPT1,可导致PC和LPC积累,并显著提高对5-FU、OXA和FOX的IC50值,获得耐药表型。 -
CHPT1过表达细胞在化疗后,表现出γ-H2AX诱导减少、PARP和caspase-3切割减弱(凋亡减弱),同时BIP表达增加、p-eIF2α/eIF2α比率降低(内质网应激适应增强)。 -
NCI-60数据集分析显示,跨癌种细胞系中CHPT1表达与5-FU耐药呈正相关。 - 临床意义
人类蛋白质图谱显示,CHPT1在结直肠癌组织中高表达,其高表达与患者总体生存率降低的趋势相关。 - 靶向治疗潜力
-
Edelfosine对耐药HT29细胞具有更强细胞毒性。 -
Edelfosine(2 μM)处理可降低PC生物合成上游酶CKα和CCTα的蛋白水平。 -
Edelfosine与FOX在HT29细胞中表现出协同作用(联合指数CI < 1),并能大幅降低达到相同疗效所需的FOX剂量。
研究总结:
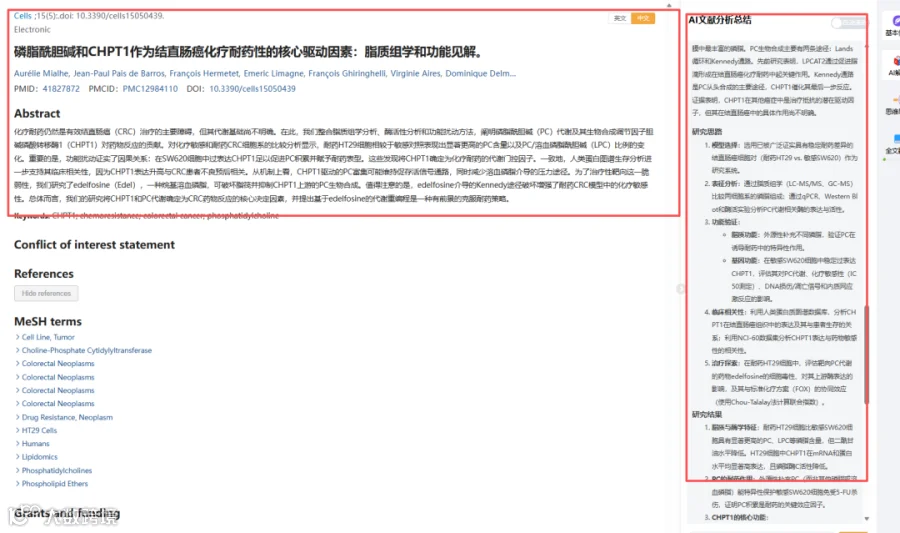
结果译文:
1.化疗敏感与耐药结直肠癌细胞的脂质组学和酶学景观:从PC重塑到磷脂酶动力学
我们首先对两种结直肠癌细胞系HT29和SW620的基底磷脂组成进行了表征。广泛的、汇聚性的证据(包括我们先前的工作[13]及多项独立研究[28-31])表明,与SW620细胞相比,HT29细胞对多种细胞毒性药物均表现出更显著的化疗耐药表型。这种可重复且有充分文献支持的差异,使HT29-SW620这对模型成为剖析化疗耐药分子决定因素的信息丰富且生物学稳健的系统。确实,针对5-氟尿嘧啶、奥沙利铂和FOLFOX的剂量-反应分析证实,HT29细胞表现出比SW620细胞更强的耐药表型,表现为曲线右移和半数抑制浓度值升高(图1A)。通过LC-MS/MS进行的基底脂质组学分析显示,HT29细胞含有更高水平的复杂磷脂和鞘脂。特别是,HT29细胞中PC、PS、LPC、LPE、LPS、神经酰胺和胆固醇的水平显著升高(图1B)。重要的是,还定量检测了DG种类,发现其在HT29细胞中相较于SW620细胞显著降低(图1B),表明磷脂周转失衡。这些观察促使我们进一步研究调控PC生物合成和水解的酶学通路(图2)。
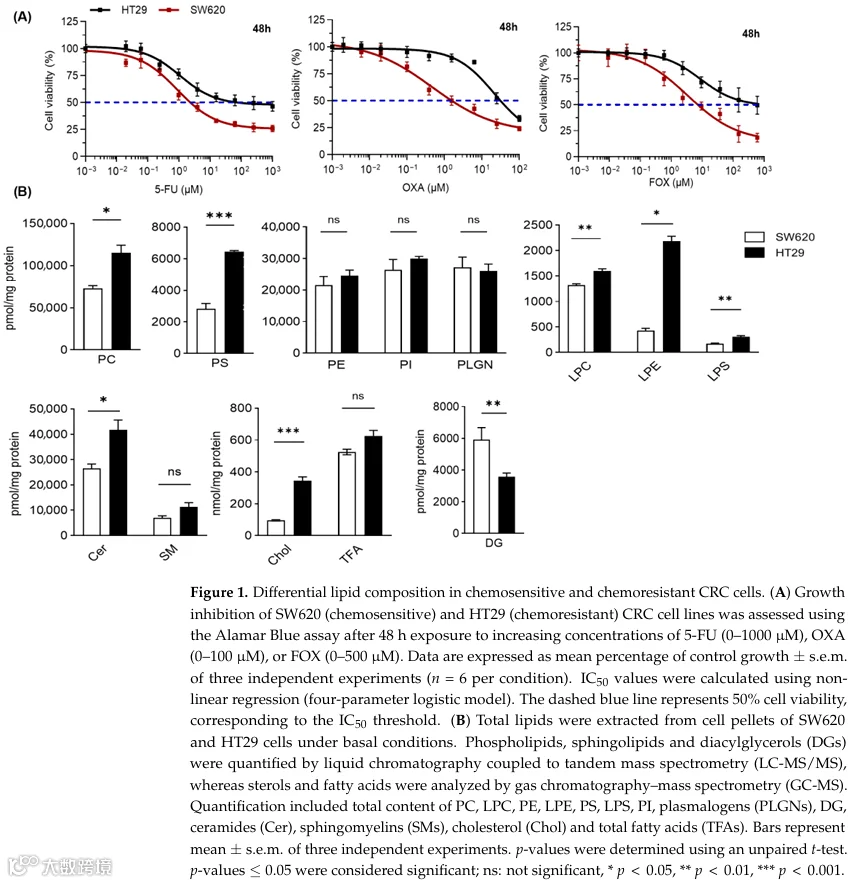
HT29细胞以高LD含量和参与PC代谢的酶(特别是LPCAT2)表达升高为特征,而SW620细胞则表现为低LD表型和LPCAT2水平降低[13]。由于从头合成PC主要受Kennedy通路控制,我们接下来评估了其关键酶CKα、CCTα和CHPT1的转录和蛋白表达,以确定该通路的差异活性是否能解释这些模型之间观察到的表型异质性(图2A)。与我们先前的工作一致,化疗耐药的HT29细胞表现出LPCAT2的显著上调,如Cotte等人[13]所述,该研究证明了LPCAT2驱动的LD生物发生支持对5-FU和OXA的耐药性(图2B、C)。值得注意的是,除了这一已确立的机制之外,我们还发现CHPT1是耐药HT29细胞系中一个显著过表达的因素,在mRNA和蛋白水平上表达均显著增加(图2B、C)。
为了进一步研究磷脂周转,我们接下来评估了参与PC水解的主要磷脂酶的活性。PC通过Kennedy途径合成,其中胆碱被CKα磷酸化,由CCTα转化为CDP-胆碱,然后由CHPT1与DG缩合形成PC。PC随后可通过Lands循环重塑,其中PLA2产生LPC和游离FA,而LPCAT酶重新酰化LPC以再生PC(图2A)。有趣的是,我们观察到化疗耐药的HT29细胞中PLC活性显著降低,而PLA2和PLD活性在敏感和耐药模型之间保持不变(图2D)。由于PLC介导的磷脂水解是细胞内DG的主要来源,PLC活性降低预计会限制DG的生成。与之一致,脂质组学分析显示HT29细胞中DG水平降低(图1B)。结合CHPT1的显著上调(其消耗DG作为PC生物合成的底物),这些发现支持PC周转的协调转变,其特征为水解减少和DG利用向PC生产增强。这种PC富集可能有助于在化疗应激下维持膜重塑和促生存信号,从而增强耐药表型。
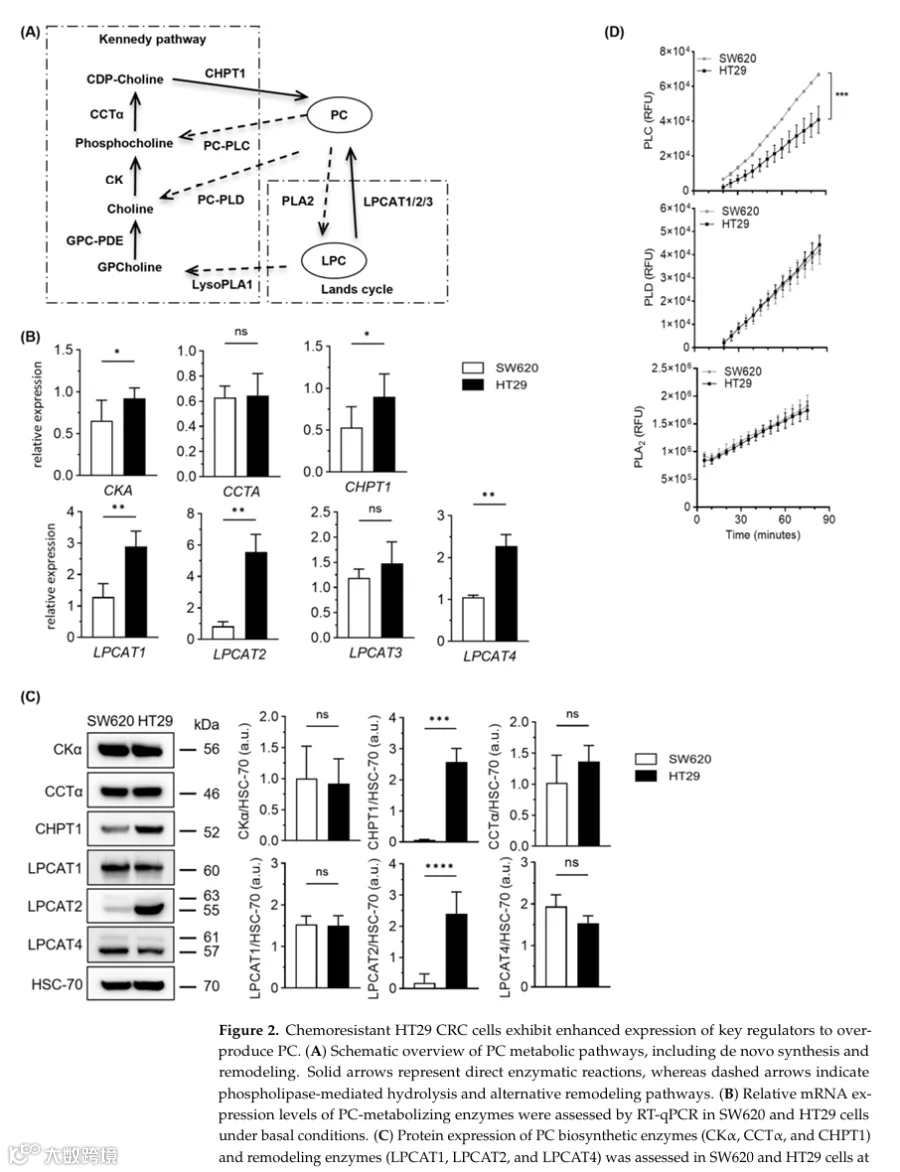
2.PC积累促进结直肠癌化疗耐药
基于我们观察到的化疗耐药HT29细胞同时具有LPCAT2上调和显著的CHPT1过表达,我们接下来探究PC本身是否是一个关键的耐药脂质决定因素。为此,我们在化疗敏感的SW620细胞中进行了外源性磷脂补充实验,引入确定量的主要磷脂及其相应的溶血磷脂,然后评估细胞对抗癌药物5-FU的反应(图3A)。重要的是,为确保有效递送和不同条件下可比较的暴露量,我们通过LC-MS/MS脂质组学分析定量了每种补充脂质掺入细胞膜的情况(图3B)。引人注目的是,只有PC补充足以重现耐药样表型,显著提高5-FU处理下的细胞活力。相比之下,补充其他磷脂或溶血磷脂在相同条件下并未赋予保护作用(图3A)。
这些功能数据支持PC富集(而非一般的磷脂可用性增加)是耐药细胞生存优势的基础这一假设。机制上,这一发现与我们之前观察到HT29细胞中PLC活性降低相一致,这可能限制PC水解,从而有利于细胞内PC积累。总之,我们的结果将PC鉴定为结直肠癌化疗耐药的关键效应脂质,并强调CHPT1驱动的PC生物合成是一个可靶向的代谢脆弱性,可用于治疗干预。
3.CHPT1是化疗反应的功能调节因子
为确定CHPT1是否直接参与化疗耐药,我们在结直肠癌模型中进行了功能获得实验。在化疗敏感的SW620细胞中稳定过表达CHPT1,并通过转录本(图4A)和蛋白(图4B)水平证实了有效诱导,与转染空载体的对照细胞相比,显示出显著增加。值得注意的是,CHPT1过表达未影响其他Kennedy通路酶(包括CKα和CCTα)的表达,支持了此操作的特异性(图4A、B)。脂质组学分析提供了机制见解,揭示了CHPT1过表达细胞中PC和LPC水平显著增加,这与PC生物合成增强和磷脂周转改变相一致(图4C)。我们接下来评估了CHPT1上调对药物反应的功能后果。使用细胞毒性测定,CHPT1过表达的SW620细胞对5-FU、OXA和联合FOX方案的IC50值显著高于对照细胞,显示出稳健的化疗耐药获得(图4D)。
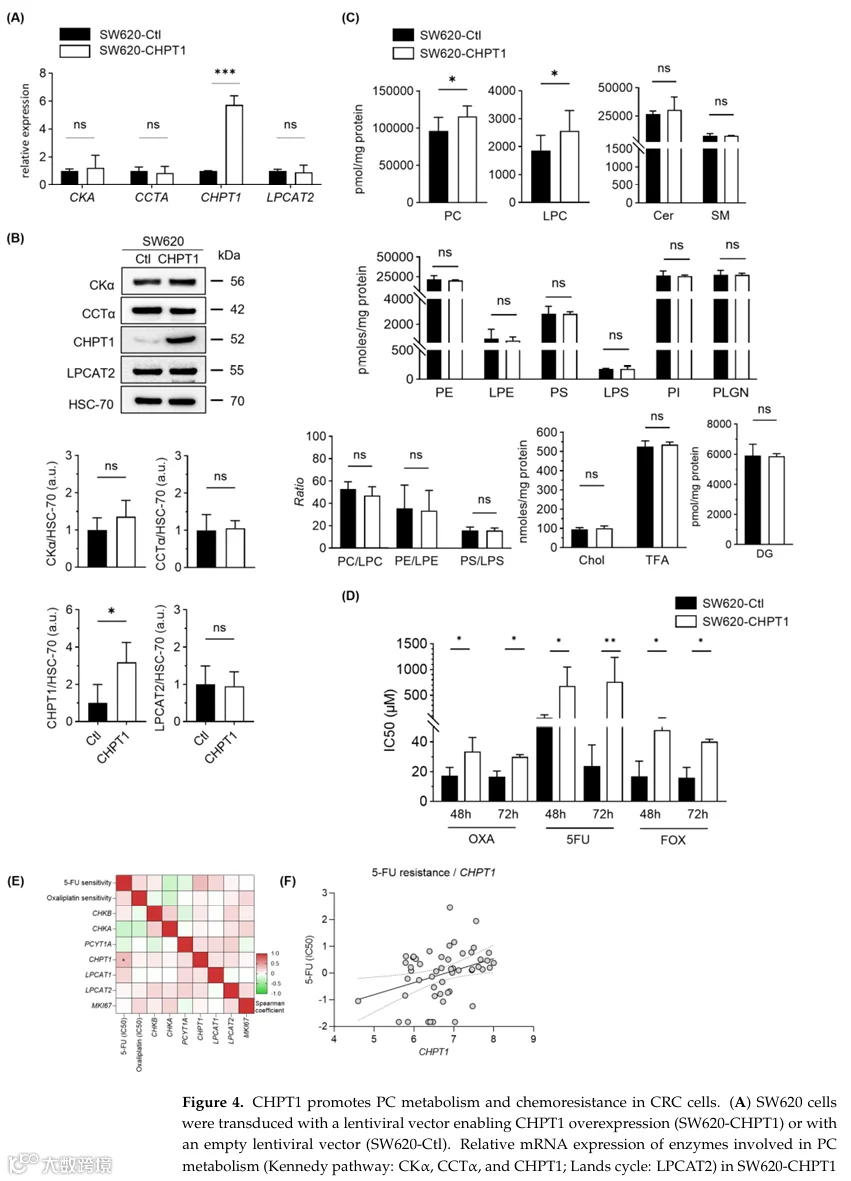

4.CHPT1表达与结直肠癌患者不良预后相关
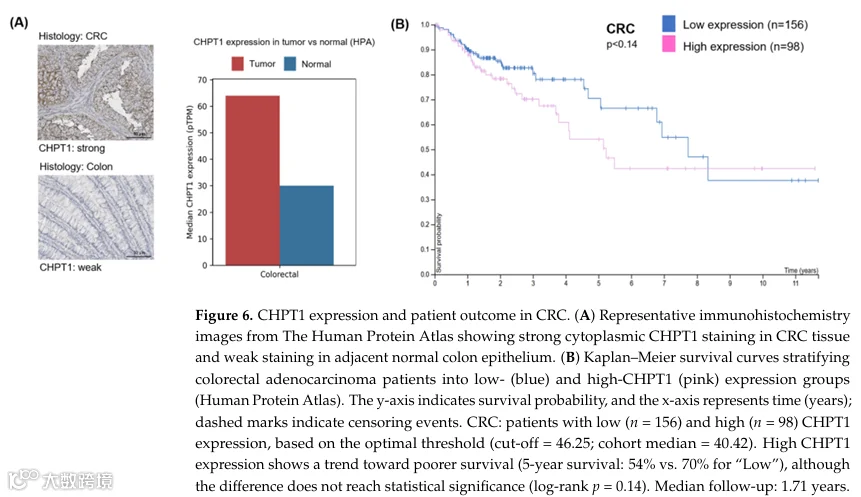
为评估CHPT1上调的临床相关性,我们分析了人类蛋白质图谱中公开的结直肠癌数据集。在结直肠癌组织中,CHPT1 mRNA表达水平为中等到高,蛋白水平验证显示与相邻正常组织相比,肿瘤细胞中表达增强。免疫组化一致显示结直肠癌样本中强烈的细胞质CHPT1染色,而相邻正常结肠上皮则表现为低表达或检测不到表达(图6A),这与CHPT1在PC生物合成中的作用及我们在化疗耐药结直肠癌细胞中的体外观察结果一致。
我们接下来检查了CHPT1表达与结直肠腺癌患者生存之间的关联。Kaplan-Meier分析显示,与低CHPT1表达患者相比,高CHPT1表达患者的总生存期呈缩短趋势(图6B)。尽管在该队列中这一差异未达到统计学显著性(对数秩p=0.14),但这些数据提示升高的CHPT1水平可能与较差的临床结局相关,并支持CHPT1作为结直肠癌候选生物标志物和治疗脆弱性的潜在相关性。
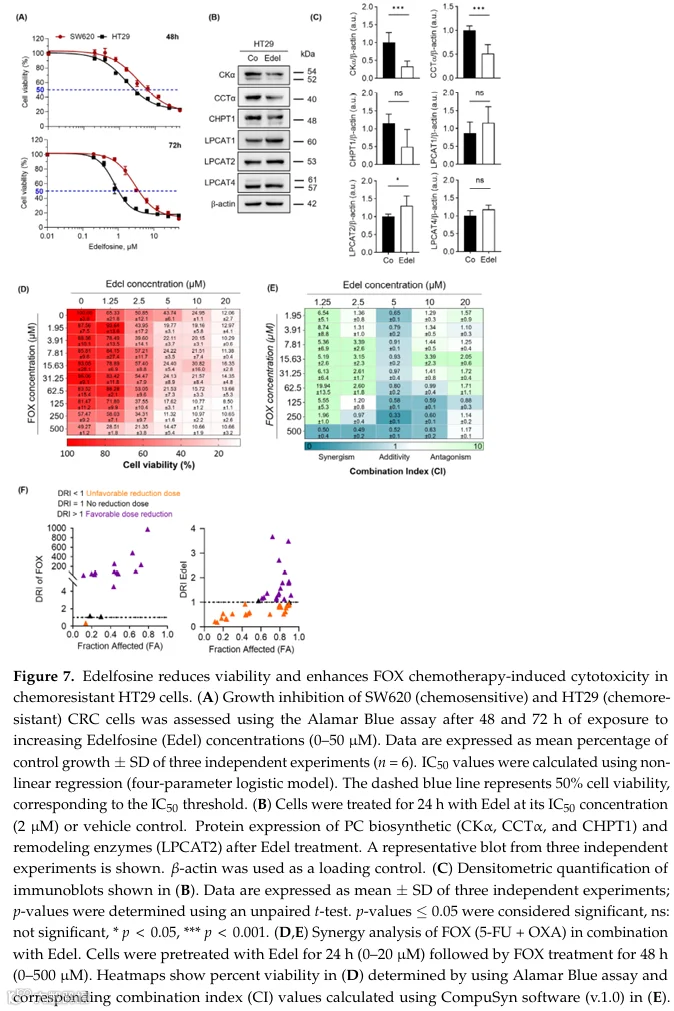
5.埃德福辛破坏PC稳态并增强化疗疗效
更多结果和补充图表:doi:10.3390/cells15050439


扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:
https://cnsknowall.com
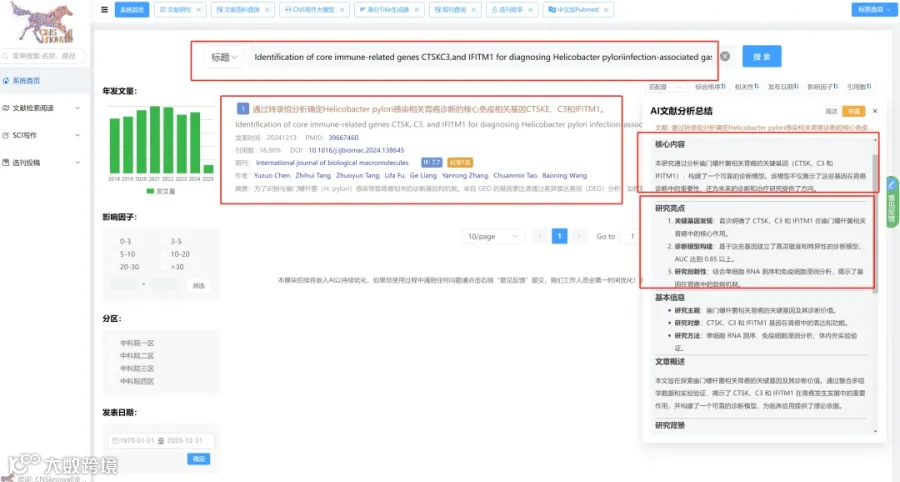
CNSknowall:24年最新问世的遥遥领先的科研数据(0代码生信+统计学)分析平台,同时含有机制图模块+汉化版Pubmed融合Deepseek高效筛选目标文献+SCI文献例句/语料检索模块+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动批阅!