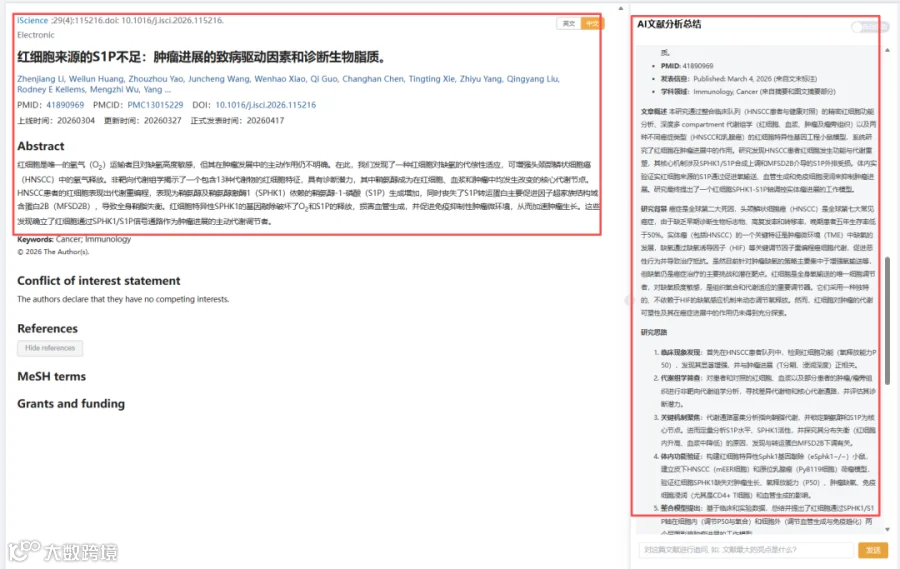
头颈部鳞状细胞癌(HNSCC)中肿瘤缺氧微环境诱导红细胞适应性反应:P50显著升高(氧释放能力增强)。非靶向代谢组学发现HNSCC患者红细胞和血浆中13个代谢物特征(AUC=0.997),鞘氨醇为核心代谢节点。机制上,HNSCC红细胞SPHK1活性升高、S1P转运蛋白MFSD2B下调,导致红细胞内S1P蓄积而血浆S1P缺乏。红细胞特异性SPHK1敲除小鼠(eSphk1-/-)肿瘤生长加速、氧释放能力下降、肿瘤缺氧加剧、CD4+ T细胞浸润减少、微血管密度降低。另一乳腺癌模型验证相似结果。该研究首次证明红细胞代谢重编程通过SPHK1-S1P-MFSD2B轴调控肿瘤血管生成与免疫浸润,为碳代谢-鞘脂代谢交叉热点提供了全新液体活检标志物(红细胞S1P相关代谢物)及治疗靶点(SPHK1/MFSD2B)。
今天给大家解读一篇3月发表在《iScience》上的题目为“Insufficient erythrocyte-derived S1P: A pathogenic driver and diagnostic biolipid for tumor progression.”的文章。本研究通过整合临床队列(HNSCC患者与健康对照)的精密红细胞功能分析、深度多 compartment 代谢组学(红细胞、血浆、肿瘤及瘤旁组织)以及两种不同癌症类型(HNSCC和乳腺癌)的红细胞特异性基因工程小鼠模型,系统研究了红细胞在肿瘤进展中的作用。研究发现HNSCC患者红细胞发生功能与代谢重塑,其核心机制涉及SPHK1/S1P合成上调和MFSD2B介导的S1P外排受损。体内实验证实红细胞来源的S1P通过促进氧输送、血管生成和免疫细胞浸润来抑制肿瘤进展。研究最终提出了一个红细胞SPHK1-S1P轴调控实体瘤进展的工作模型。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!包括数据集、绘图代码、图表复现、思路总结、参考文献……0代码!鼠标点点点即可轻松完成5-10分生信SCI全文复现!
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!



团队成员合影(位于上海陆家嘴中心,可随时预约参观)
题目:《红细胞来源的S1P不足:肿瘤进展的致病驱动因素和诊断生物脂质》Insufficient erythrocyte-derived S1P: A pathogenic driver and diagnostic biolipid for tumor progression
发表期刊:iScience
影响因子:4.1
研究背景:
癌症是全球第二大死因,头颈鳞状细胞癌(HNSCC)是全球第七大常见癌症,由于缺乏早期诊断生物标志物、高复发率和转移率,晚期患者五年生存率低于50%。实体瘤(包括HNSCC)的一个关键特征是肿瘤微环境(TME)中缺氧的发展,缺氧通过缺氧诱导因子(HIF)等关键调节因子重编程癌细胞代谢,促进恶性行为并导致治疗抵抗。虽然目前针对肿瘤缺氧的策略主要集中于增强氧输送等,但缺氧仍是癌症治疗的主要挑战和潜在靶点。红细胞是全身氧输送的唯一细胞调节者,对缺氧极度敏感,是组织氧合和代谢适应的重要调节器。它们采用一种独特的、不依赖于HIF的缺氧感应机制来动态调节氧释放。然而,红细胞对肿瘤的代谢可塑性及其在癌症进展中的作用仍未得到充分探索。
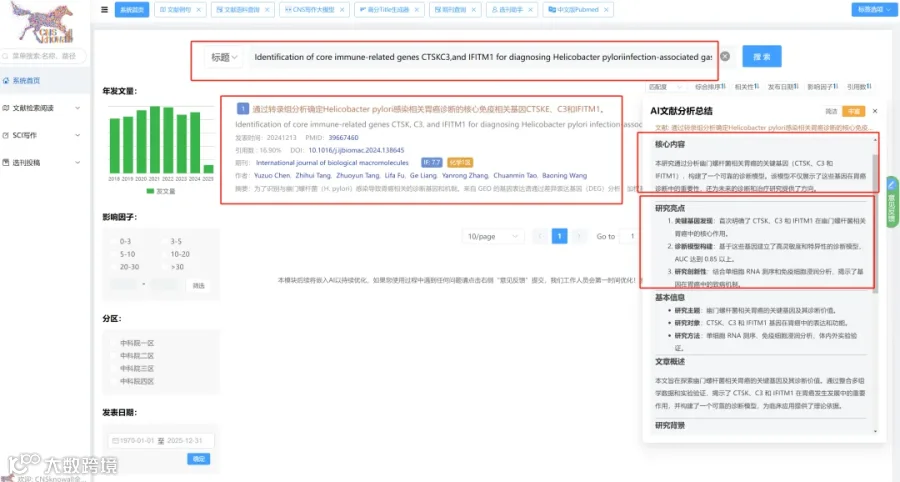
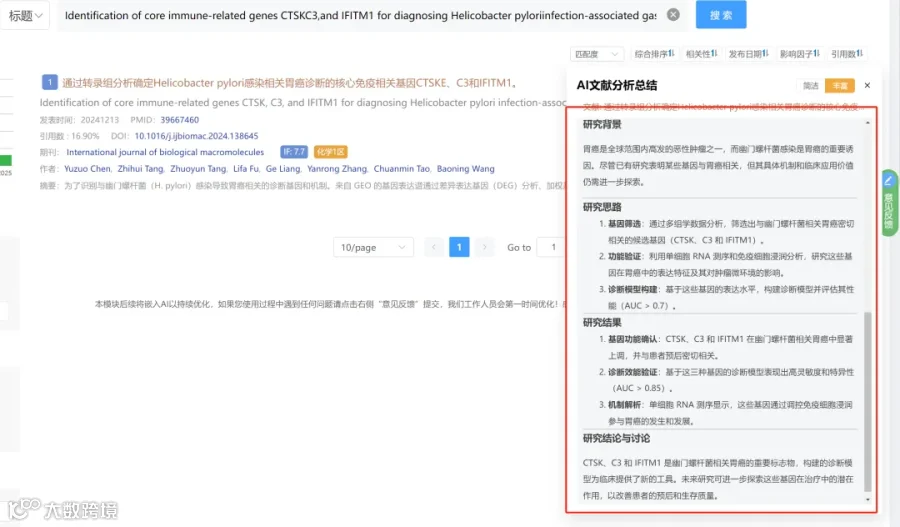
CNSknowall 平台 Pubmed+AI 快速提炼全文要点
研究思路:
- 临床现象发现
首先在HNSCC患者队列中,检测红细胞功能(氧释放能力P50),发现其显著增强,并与肿瘤进展(T分期、浸润深度)正相关。
- 代谢组学筛查
对患者和对照的红细胞、血浆以及部分患者的肿瘤/瘤旁组织进行非靶向代谢组学分析,寻找差异代谢物和核心代谢通路,并评估其诊断潜力。
- 关键机制聚焦
代谢通路富集分析指向鞘脂代谢,并锁定鞘氨醇和S1P为核心节点。进而定量分析S1P水平、SPHK1活性,并探究其分布失衡(红细胞内升高、血浆中降低)的原因,发现与转运蛋白MFSD2B下调有关。
- 体内功能验证
构建红细胞特异性Sphk1基因敲除(eSphk1−/−)小鼠,建立皮下HNSCC(mEER细胞)和原位乳腺癌(Py8119细胞)荷瘤模型,验证红细胞SPHK1缺失对肿瘤生长、氧释放能力(P50)、肿瘤缺氧、免疫细胞浸润(尤其是CD4+ T细胞)和血管生成的影响。
- 整合模型提出
基于临床和实验数据,总结并提出了红细胞通过SPHK1/S1P轴在细胞内(调节P50与氧合)和细胞外(调节血管生成与免疫趋化)两个层面影响肿瘤进展的工作模型。
研究亮点:
-
-
红细胞-血浆鞘脂失衡建立了一个促肿瘤的系统性生态位。
-
红细胞SPHK1缺失通过破坏血管生成和免疫浸润促进肿瘤生长。
-
研究整合了人类队列的代谢组学分析与基因工程小鼠模型,提供了机制性证据。
研究结果:
- 红细胞功能改变
HNSCC患者红细胞的P50值显著高于健康对照,表明其氧释放能力增强,且P50值与肿瘤T分期和浸润深度呈正相关。
- 代谢重编程与诊断标志物
红细胞和血浆代谢谱在HNSCC与对照组间存在显著差异。鉴定出13种共有的差异代谢物(包括乳酸和鞘氨醇),其组合在区分HNSCC与对照时ROC曲线下面积(AUC)达0.997。这些代谢物在肿瘤进展中呈现三种动态变化模式。
- 鞘脂代谢失衡机制
鞘脂代谢是红细胞、血浆和肿瘤组织中最显著富集的通路。鞘氨醇是三个 compartment 中唯一共同且变化最显著的代谢物。HNSCC患者红细胞中鞘氨醇、S1P水平及SPHK1活性均显著升高,但血浆S1P水平却显著降低。这种倒置关系与红细胞膜上S1P转运蛋白MFSD2B的表达显著下调相关。
- 红细胞SPHK1/S1P的抑瘤功能验证
- 肿瘤生长
在eSphk1−/−小鼠的HNSCC和乳腺癌模型中,肿瘤生长更快、重量更大。
- 氧合与缺氧
eSphk1−/−小鼠红细胞SPHK1活性和P50值均降低,肿瘤组织缺氧加重。
- 免疫微环境
eSphk1−/−小鼠肿瘤内CD45+免疫细胞(在HNSCC模型中主要为CD4+ T细胞)浸润显著减少。
- 血管生成
eSphk1−/−小鼠肿瘤内微血管密度(CD31+)显著降低。
- S1P分布
eSphk1−/−荷瘤小鼠红细胞和血浆S1P水平均降低,但肿瘤组织内S1P水平无显著变化。
研究总结:
本研究结论是,红细胞是肿瘤进展的主动代谢调节器。HNSCC中,红细胞发生代谢重编程,通过上调SPHK1活性增强S1P合成,但同时因MFSD2B下调导致S1P外排受阻,形成“红细胞内S1P蓄积-血浆S1P耗竭”的系统性失衡。红细胞来源的S1P通过双重机制抑制肿瘤:细胞内,S1P增强红细胞氧释放能力(提高P50),缓解肿瘤缺氧;细胞外,释放至血浆的S1P促进肿瘤血管生成和免疫细胞(特别是T细胞)向肿瘤微环境的趋化。
讨论指出,红细胞P50升高可能是对肿瘤缺氧的补偿反应,也可作为疾病严重程度的间接指标和潜在生物标志物。研究强调了红细胞代谢特征作为微创诊断工具的潜力。同时,研究澄清了红细胞来源的SPHK1/S1P轴具有抑瘤作用,这与多数关于肿瘤细胞自身SPHK1促瘤作用的研究不同,提示需要区分不同细胞来源S1P的功能。文末提出了一个整合性的工作模型,并指出未来需进一步阐明S1P与特定受体在TME中相互作用的分子机制,以及利用患者来源类器官等模型验证红细胞与肿瘤细胞的直接相互作用。
结果译文:
1.头颈部鳞状细胞癌患者红细胞氧释放能力增强且与肿瘤进展密切相关
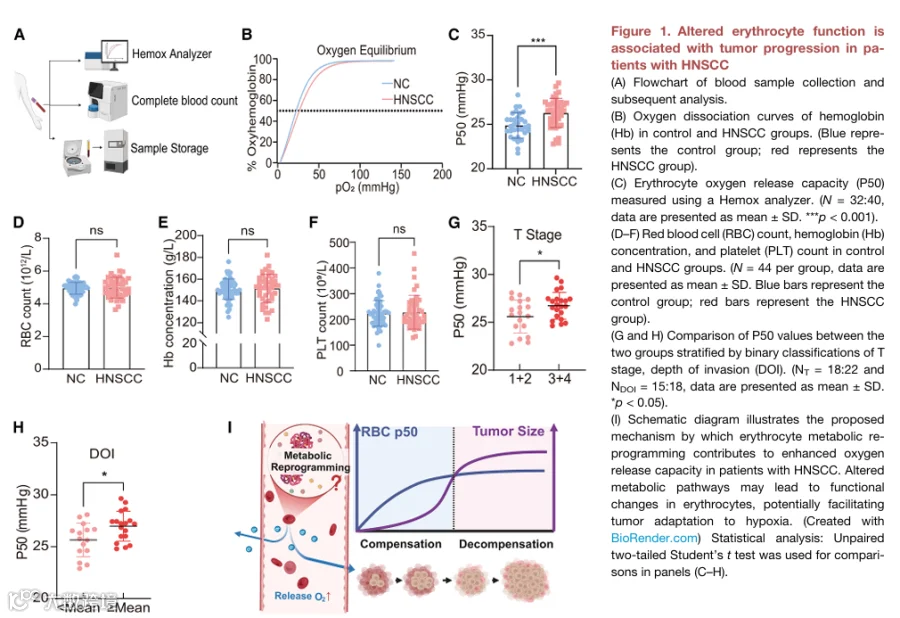
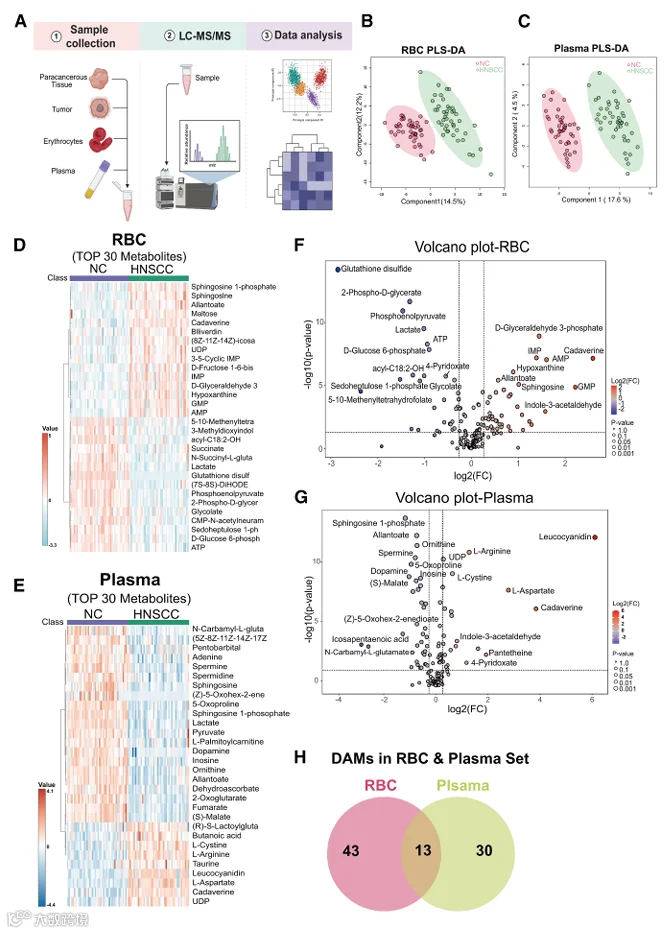
为了研究HNSCC患者红细胞的功能改变并评估其在早期诊断中的潜在应用价值,我们纳入了一个特征明确的人类队列,包括HNSCC患者和健康志愿者(表1)。从两组中收集外周血样本(图1A)。血红蛋白50%饱和时的氧张力反映了红细胞的氧释放能力。为了探索HNSCC患者红细胞的功能变化,我们使用血氧分析仪测量了HNSCC患者与健康对照红细胞样本之间P50的差异。结果显示,HNSCC红细胞的P50显著高于健康对照(图1B和1C),精确定义了HNSCC患者中血红蛋白-氧亲和力降低和氧释放增强。
贫血可能影响血氧测定法获得的P50测量值。为了排除红细胞和血红蛋白浓度对P50值的影响并比较炎症指标,我们对两组进行了全血细胞计数分析。CBC分析显示,两组之间的红细胞计数(图1D)、血红蛋白浓度(图1E)或血小板计数(图1F)无显著差异。这些发现表明,观察到的P50变化不太可能主要归因于红细胞数量或寿命的差异。
接下来,我们通过根据T分期(T1+T2 vs. T3+T4)对患者进行分层,进行了P50与肿瘤进展之间的相关性分析。我们发现,晚期疾病患者(T3+T4)表现出显著更高的P50值(图1G)。类似地,当根据平均浸润深度对患者进行分组时,浸润深度较高的患者也表现出升高的P50值(图1H)。这些发现进一步支持了红细胞氧释放能力随肿瘤进展而增强的观点。
2.红细胞代谢重编程反映HNSCC疾病进展并可作为诊断生物标志物
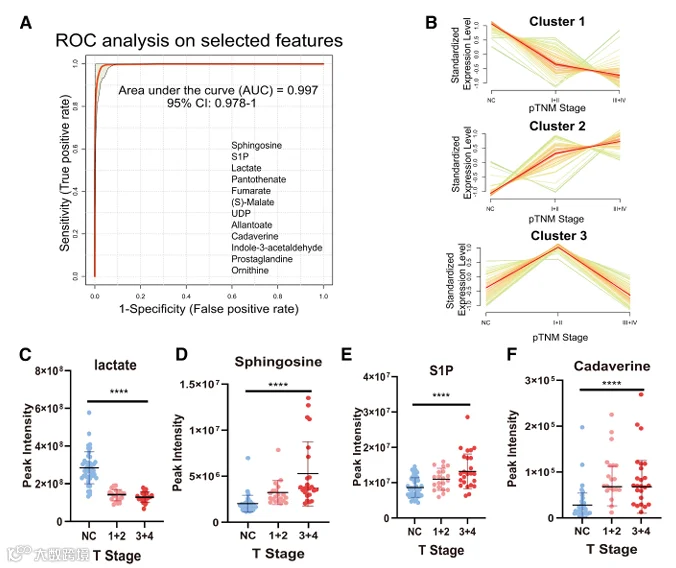
鉴于红细胞功能与代谢重编程之间的已知联系,我们假设在HNSCC期间红细胞中发生代谢改变,导致观察到的P50变化(图1I)。因此,为了研究与HNSCC相关的红细胞代谢改变,我们收集了44对HNSCC患者和匹配健康对照的红细胞和血浆样本(图1A),以及13例HNSCC患者的肿瘤和癌旁组织(图2A)。然后对所有样本进行了非靶向代谢组学分析。最初,为了寻找潜在的循环差异代谢谱,分别基于红细胞和血浆代谢谱进行了偏最小二乘判别分析。我们在红细胞和血浆中均鉴定出HNSCC组和对照组之间的明显分离(图2B和2C)。热图和火山图分析在两个区室中均鉴定出两组之间的大量差异丰度代谢物(图2D-2G)。接下来,来自红细胞和血浆数据集的DAMs的交集揭示了13个共享代谢物,包括乳酸和鞘氨醇(图2H)。为了评估这些代谢物的联合诊断潜力,我们基于13个共享代谢物的整合代谢谱进行了受试者工作特征(ROC)分析。所有选定代谢物的曲线下面积(AUC)值为0.997,表明区分HNSCC与健康对照具有高预测准确性(图3A)。
此外,为了探索这些代谢物在HNSCC进展过程中的动态变化,我们进行了Mfuzz聚类。出现了三种不同的模式:簇1代谢物表现出持续下降,簇2代谢物显示逐渐积累,簇3代谢物表现出双相模式,先升高后下降(图3B)。这些模式通过代表性代谢物说明(图3C-3F):乳酸(簇1)、鞘氨醇和S1P(簇2)以及尸胺(簇3)。总之,这些发现表明,在肿瘤进展过程中,红细胞经历显著的代谢重编程。红细胞和血浆的代谢谱不仅反映了肿瘤进展的状态,而且有潜力作为HNSCC进展的预测性生物标志物。
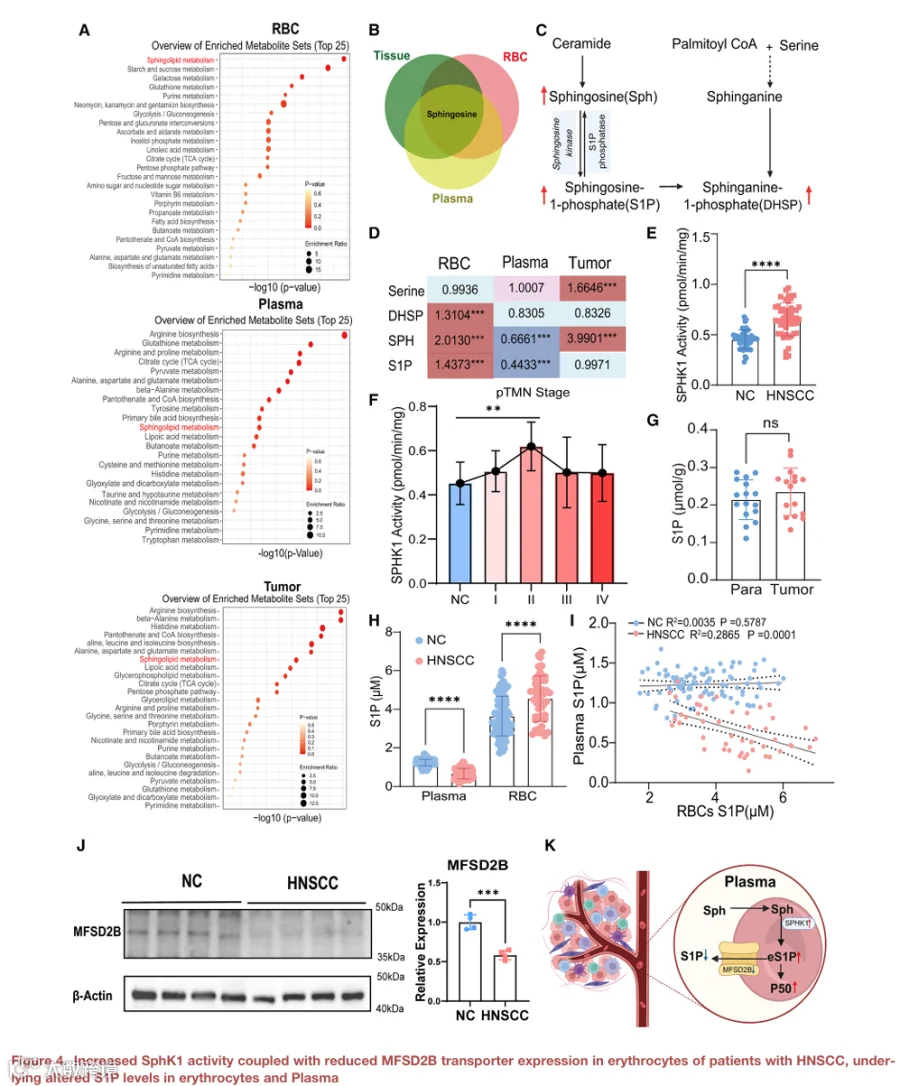
3.HNSCC中红细胞鞘氨醇和SPHK1活性增加,同时S1P转运蛋白MFSD2B下调,导致鞘脂失衡
为了研究红细胞代谢重编程与HNSCC进展之间的关系,我们进行了京都基因与基因组百科全书(KEGG)通路富集分析。鞘脂代谢在红细胞、血浆和肿瘤组织中富集最显著(图4A)。值得注意的是,鞘氨醇——一种关键的鞘脂中间体——被鉴定为基于液相色谱-质谱(LC-MS)分析在所有三个区室中唯一常见且变化最显著的代谢物(图4B)。鉴于红细胞通过S1P相关通路适应缺氧的既定作用,我们专门检查了非靶向代谢组学数据集中S1P相关的代谢变化。结果显示,HNSCC红细胞中鞘氨醇和S1P的水平均显著升高(图4C和4D)。SPHK1是红细胞中负责S1P产生的主要酶。为了评估HNSCC患者和健康对照红细胞中SPHK1的酶活性,我们进行了活性测定,发现HNSCC组中SPHK1活性显著升高(图4E)。此外,根据病理TNM分期进行分层显示,SPHK1活性在II期患者中最高(图4F)。靶向LC-MS定量证实了这些发现。令我们惊讶的是,虽然S1P水平在肿瘤和癌旁组织之间没有显著差异(图4G),但HNSCC患者的红细胞显示出显著更高的S1P水平;相反,与对照组相比,血浆S1P显著降低(图4H)。相关性分析进一步支持了这种反比关系,显示HNSCC组中红细胞和血浆S1P水平之间存在强烈的负相关,而这种模式在对照组中不存在(图4I)。因此,这些发现揭示,SPHK1活性升高伴随鞘氨醇增加是红细胞中S1P水平升高的基础,并且是肿瘤进展期间氧释放增加的潜在分子机制。
由于大约50%的血浆S1P来源于红细胞,我们假设S1P输出受损可能导致观察到的血浆耗竭。MFSD2B是红细胞中主要的S1P转运蛋白,先前被确定为这一过程的关键参与者。支持这种可能性的是,蛋白质印迹分析显示,与对照组相比,HNSCC患者的红细胞中MFSD2B的表达显著降低(图4J)。因此,我们发现红细胞中MFSD2B的下调似乎损害了S1P的外排,导致HNSCC中血浆S1P缺乏(图4K)。这种失调可能影响关键的肿瘤相关过程,包括血管生成和免疫细胞运输,从而促进HNSCC的发病机制。
4.红细胞来源的S1P通过促进TME中的氧输送、血管生成和免疫细胞浸润来对抗肿瘤进展
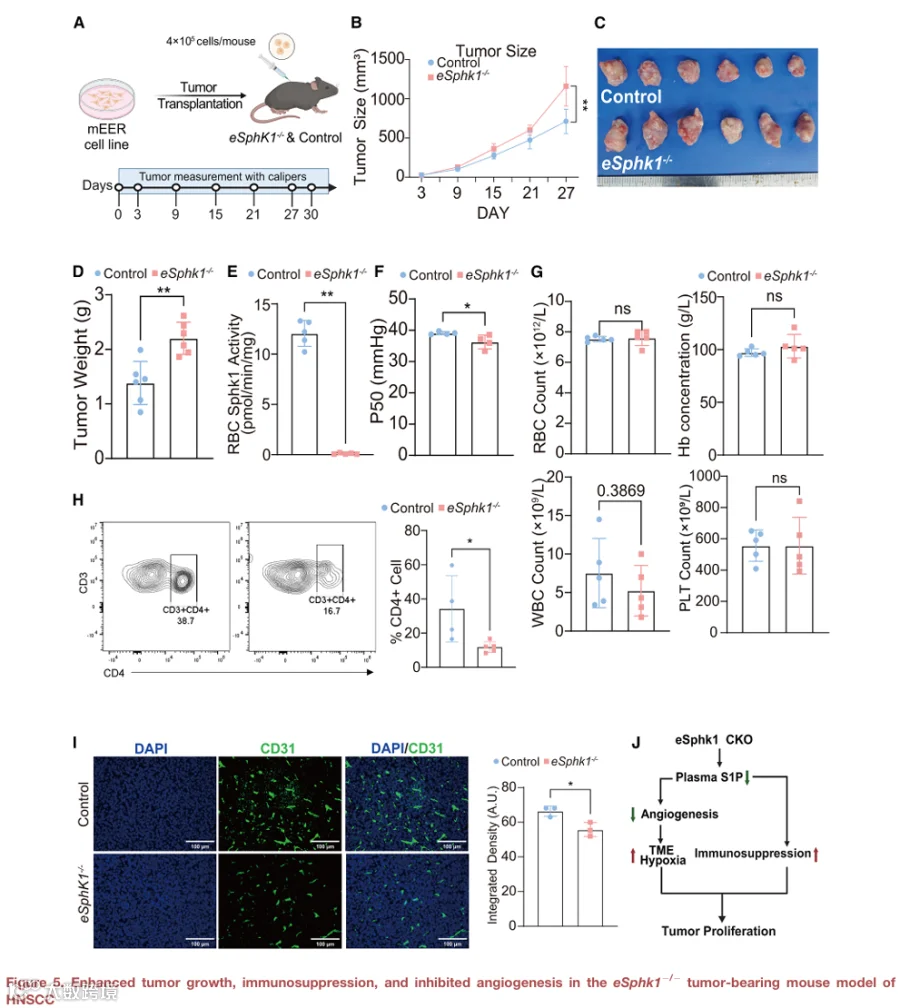
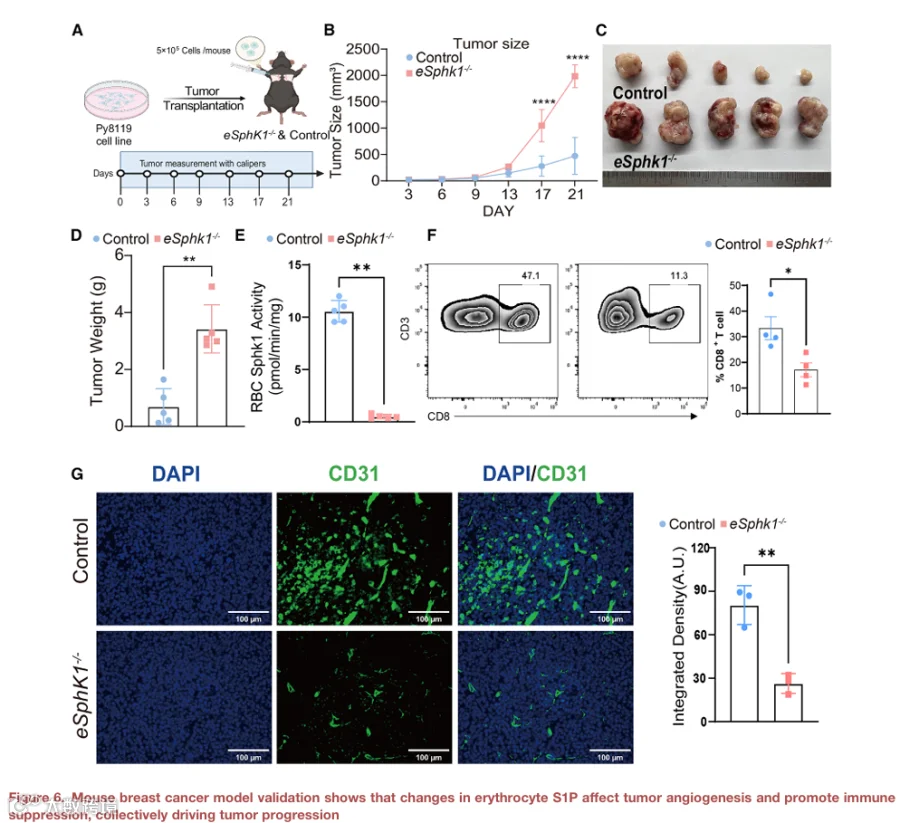
确定红细胞来源S1P的减少是否促进人类肿瘤进展具有挑战性。为了解决这个问题,我们使用小鼠模型进行了体内实验。具体来说,我们在eSphk1-/-小鼠和对照小鼠(Sphk1fl/fl)中建立了皮下HNSCC荷瘤模型(图5A)。我们的观察显示,与对照小鼠相比,eSphk1-/-小鼠中的肿瘤生长显著更快且重量更大(图5B-5D),表明eSphk1在对抗肿瘤发展中发挥重要作用。
为了明确eSphk1在肿瘤进展中发挥有益作用的分子基础,我们首先测量了SPHK1酶活性和P50值。敲除组中两者均显著降低(图5E和5F),表明eSphk1对于响应肿瘤发展促进氧释放能力至关重要。接下来,我们评估了红细胞SPHK1-S1P缺乏对肿瘤缺氧的影响,使用hypoxyprobe(哌莫硝唑,在缺氧细胞中形成蛋白加合物)进行评估。hypoxyprobe染色结果显示,与对照小鼠相比,eSphk1-/-小鼠携带的肿瘤具有显著降低的氧合水平,表明瘤内缺氧增加(图S1)。
为了表征eSphk1-/-对荷瘤模型中免疫调节的影响,我们对肿瘤组织进行了流式细胞术分析和免疫荧光染色。结果显示,eSphk1-/-组中瘤内CD4+ T细胞浸润显著减少(图5H),表明免疫细胞趋化性受损,这可能削弱抗肿瘤免疫应答。鉴于S1P在血管生成中的重要性,我们通过CD31免疫荧光染色对这两组的血管生成进行了定量。我们发现,与对照组相比,eSphk1-/-组肿瘤中的微血管密度降低(图5I)。
为了定量定义红细胞来源的SPHK1对全身和肿瘤相关鞘氨醇-1-磷酸(S1P)池的贡献,我们对对照和eSphk1-/-小鼠的红细胞、血浆和肿瘤组织中的S1P水平进行了靶向质谱分析。在荷瘤小鼠中,与对照同窝小鼠相比,eSphk1-/-小鼠的红细胞S1P水平显著降低,其循环血浆S1P也相应减少(图S2)。这表明红细胞中SPHK1的缺失影响红细胞内和血浆中的S1P水平。相比之下,定量分析显示,eSphk1-/-和对照小鼠之间的mEER肿瘤组织内S1P水平无显著差异。这表明,尽管红细胞来源的S1P缺失,肿瘤内在的S1P仍然基本保持完整,表明观察到的表型差异不太可能由肿瘤细胞来源的S1P变化驱动。
总之,我们提供了遗传学概念验证证据,表明红细胞SPHK1通过促进O2和S1P释放来对抗HNSCC肿瘤进展,从而增强肿瘤微环境中的氧合、血管生成以及免疫细胞浸润(图5J)。
最后,为了确定eSphk1在肿瘤进展中的广泛保护作用,我们使用Py8119细胞建立了独立的原位肿瘤移植模型,以模拟其天然微环境中的乳腺癌生长(图6A)。类似地,我们发现eSphk1缺失导致严重的肿瘤进展,表现为肿瘤尺寸大、血管生成减少和免疫抑制增强(图6B-6G)。总之,这些结果表明,红细胞来源S1P的减少导致免疫抑制和血管生成缺陷的微环境,从而促进HNSCC进展。
更多结果和补充图表:doi: 10.1016/j.isci.2026.115216
长按二维码关注我们,用最短的时间和最高的效率学习更多数据分析方法!
扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:
https://cnsknowall.com
CNSknowall:24年最新问世的遥遥领先的科研数据(0代码生信+统计学)分析平台,同时含有机制图模块+汉化版Pubmed融合Deepseek高效筛选目标文献+SCI文献例句/语料检索模块+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动批阅!