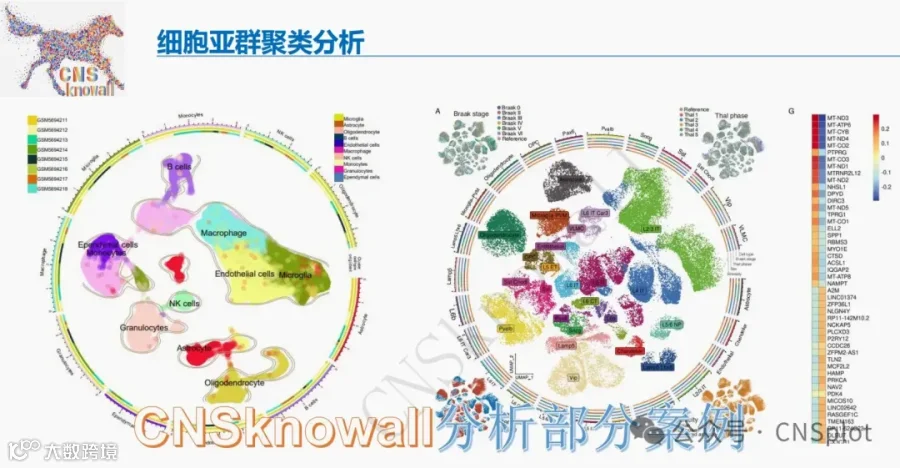
重磅!体内CRISPR筛选发现表观遗传免疫检查点CBX4!CBX4是PRC1复合物组分,本研究通过体内CRISPR-Cas9筛选(靶向998个表观遗传基因)在肝癌和结肠癌模型中发现:CBX4缺失显著增敏抗PD-1治疗。单细胞转录组和空间转录组显示,CBX4在肿瘤细胞和免疫抑制性TAMs中高表达,与免疫治疗耐药相关。机制上,CBX4通过H3K9me3/H3K27me3修饰沉默内源逆转录元件(ERV)RLTR4-Mm-int;CBX4缺失后ERV去抑制,胞质dsRNA积累,激活RIG-I-MAVS通路,触发I型干扰素应答,重塑炎性TME,增强CD8+ T和NK细胞浸润及杀伤功能。CBX4抑制剂UNC3866联合抗PD-1在PDX模型中显著抑制肿瘤。该研究为表观遗传-神经免疫交叉热点提供了全新靶点,并为ERV激动剂作为免疫增敏剂提供了理论依据!
今天给大家解读一篇3月发表在《The Journal of Clinical Investigation》上的题目为“In vivo CRISPR screens identify CBX4 as an epigenetic regulator for cancer immunotherapy.”的文章。本文报道了一项基于体内CRISPR筛选的研究,发现CBX4是免疫肿瘤微环境的关键负调节因子。通过多组学分析和机制实验,证明CBX4缺失可通过激活逆转座子介导的I型干扰素反应,增强CD8+ T细胞和NK细胞浸润及活性,从而提高ICB疗效;临床数据进一步支持CBX4作为肝细胞癌免疫治疗预后不良的标志物和潜在治疗靶点。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!包括数据集、绘图代码、图表复现、思路总结、参考文献……0代码!鼠标点点点即可轻松完成5-10分生信SCI全文复现!
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!


团队成员合影(位于上海陆家嘴中心,可随时预约参观)
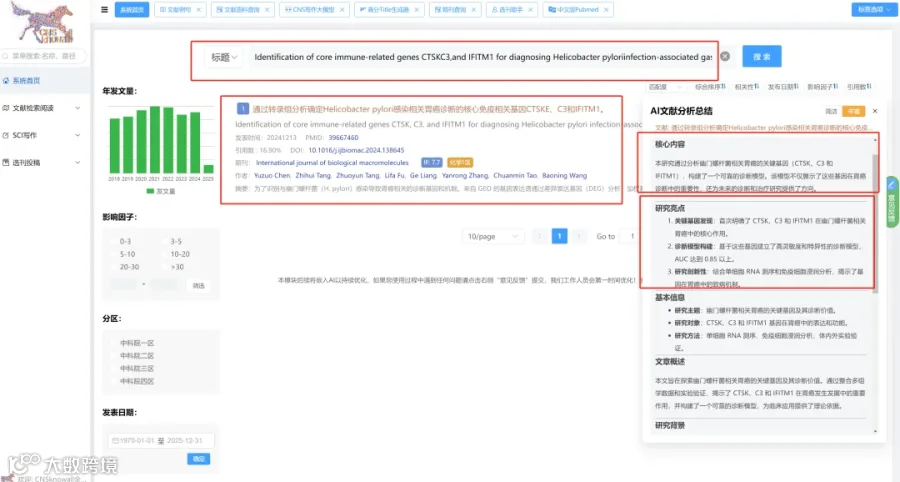
题目:《体内CRISPR筛选鉴定出CBX4作为癌症免疫治疗的表观遗传调节因子》In vivo CRISPR screens identify CBX4 as an epigenetic regulator for cancer immunotherapy
发表期刊:The Journal of Clinical Investigation
影响因子:13.6
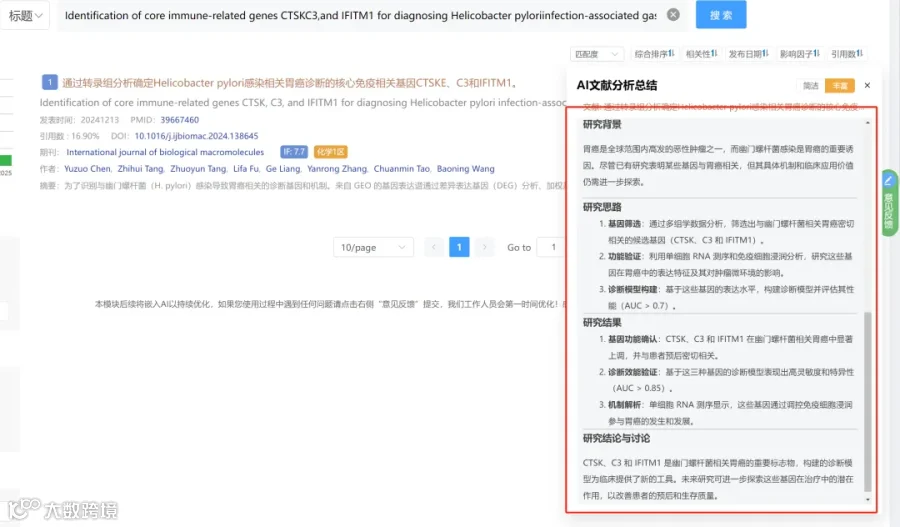
研究背景:
表观遗传失调与免疫逃逸和免疫检查点阻断(ICB)耐药相关。
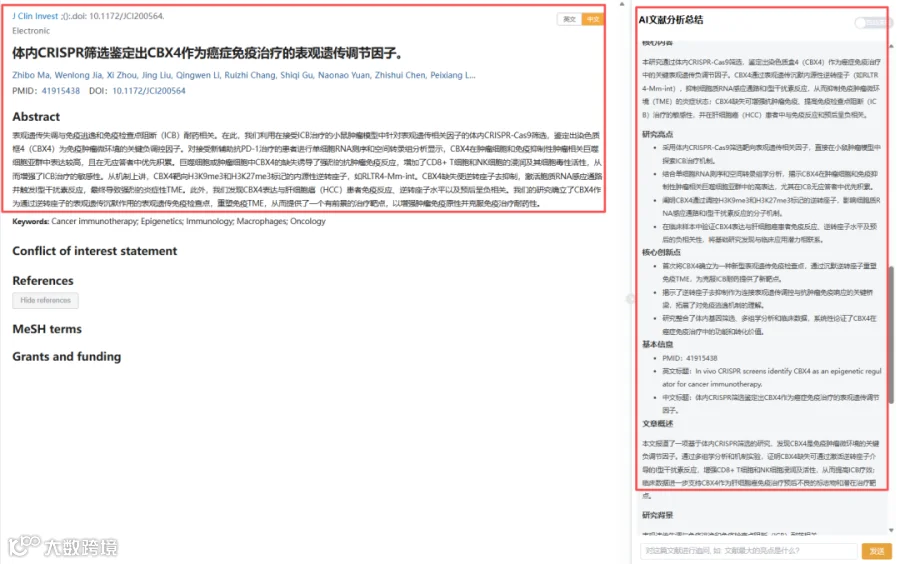
CNSknowall 平台 Pubmed+AI 快速提炼全文要点
研究思路:
-
使用体内CRISPR-Cas9筛选,在小鼠肿瘤模型(接受ICB治疗)中靶向表观遗传相关因子,以鉴定调控免疫TME的关键基因。
-
对接受新辅助抗PD-1治疗的患者样本进行单细胞RNA测序和空间转录组学分析,评估CBX4表达与治疗响应的关联。
-
通过基因缺失实验(在巨噬细胞或肿瘤细胞中),探究CBX4对抗肿瘤免疫和ICB敏感性的功能影响。
-
机制上,分析CBX4对H3K9me3和H3K27me3标记内源性逆转座子的靶向作用,及其对细胞质RNA感应通路和I型干扰素反应的调控。
-
在肝细胞癌患者队列中,分析CBX4表达与免疫响应、逆转座子水平及预后的相关性。
研究亮点:
-
采用体内CRISPR-Cas9筛选靶向表观遗传相关因子,直接在小鼠肿瘤模型中探索ICB治疗机制。
-
结合单细胞RNA测序和空间转录组学分析,揭示CBX4在肿瘤细胞和免疫抑制性肿瘤相关巨噬细胞亚群中的高表达,尤其在ICB无应答者中优先积累。
-
阐明CBX4通过调控H3K9me3和H3K27me3标记的逆转座子,影响细胞质RNA感应通路和I型干扰素反应的分子机制。
-
在临床样本中验证CBX4表达与肝细胞癌患者免疫反应、逆转座子水平及预后的负相关性,将基础研究发现与临床应用潜力相联系。
研究结果:
-
体内CRISPR筛选鉴定出CBX4为免疫TME的关键负调节因子。
-
单细胞和空间转录组学显示:CBX4在肿瘤细胞和免疫抑制性肿瘤相关巨噬细胞亚群中高表达,且在ICB无应答者中优先积累。
-
功能实验表明:CBX4在巨噬细胞或肿瘤细胞中的缺失,可诱导强抗肿瘤免疫,增加CD8+ T细胞和NK细胞的浸润及细胞毒性活性,从而增强ICB治疗敏感性。
-
机制解析发现:CBX4靶向H3K9me3和H3K27me3标记的内源性逆转座子(如RLTR4-Mm-int);CBX4缺失导致逆转座子去抑制,激活细胞质RNA感应通路并触发I型干扰素反应,最终形成炎症性TME。
-
临床相关性分析:在肝细胞癌患者中,CBX4表达与免疫响应、逆转座子水平及预后呈负相关。
研究总结:
本研究结论认为,CBX4通过表观遗传沉默逆转座子,发挥表观遗传免疫检查点的作用,抑制免疫TME的炎症状态,从而导致ICB耐药。CBX4缺失可重塑免疫TME,增强肿瘤免疫原性,为克服免疫治疗耐药提供了有前景的治疗靶点。讨论强调,这一发现揭示了表观遗传调控与先天免疫响应之间的新联系,并支持CBX4作为癌症免疫治疗中新的生物标志物和干预策略。
结果译文:
1.体内CRISPR筛选鉴定CBX4为免疫逃逸的表观遗传调节因子
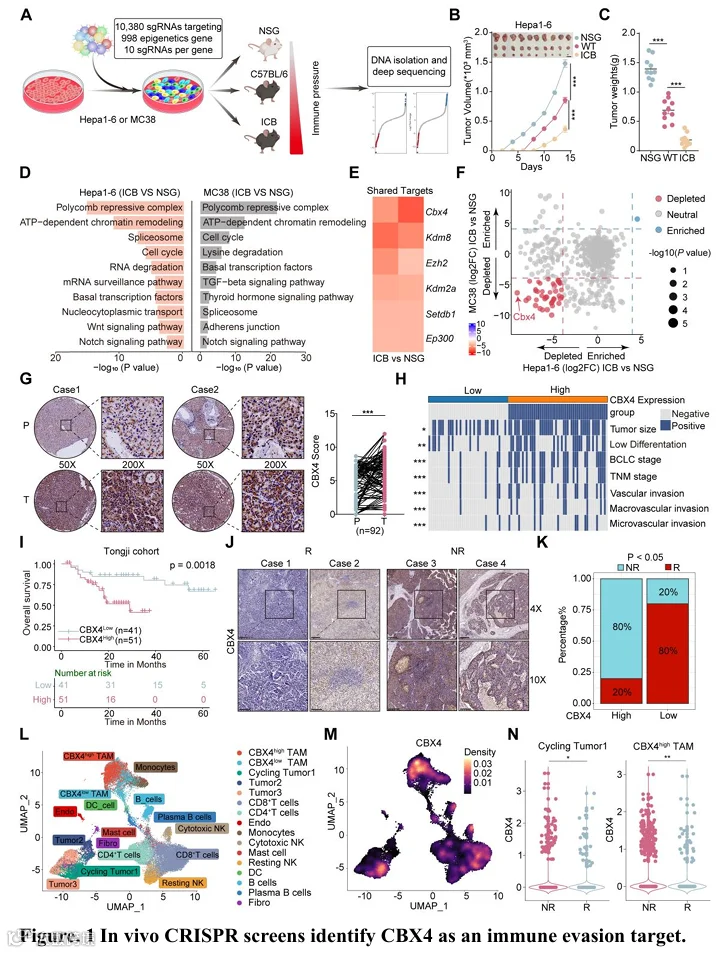
为了系统性地鉴定肿瘤细胞中表观遗传相关的免疫逃逸靶点,我们设计了一个慢病毒CRISPR-Cas9筛选文库,包含针对998个表观遗传相关基因的10个sgRNA(以达到足够的覆盖度和高靶向效率,表S1),并将其转导到两种小鼠肿瘤细胞系——Hepa1-6(肝细胞癌)和MC38(结肠癌)中。将转导了sgRNA文库的肿瘤细胞移植到免疫健全野生型小鼠、ICB治疗的野生型小鼠以及作为对照的免疫缺陷NOD SCID II2rg-(NSG)小鼠中,以鉴定具有免疫依赖性效应的sgRNA(图1A)。收集肿瘤后,我们进行了文库质量控制,并观察到由内源性抗肿瘤免疫或ICB治疗驱动的肿瘤生长抑制(图1B、1C和S1A-D;表S1)。除了鉴定出先前文库研究中发现的与ICB耐药相关的基因(如Ezh2、Setdb1、Ep300和Mettl14)外,在免疫压力下,靶向多梳抑制复合物通路的sgRNA在肿瘤细胞中显著耗竭(图1D-E,S1E-G)。在验证实验中,在荷Hepa1-6肿瘤小鼠中,Cbx4、Kdm8、Ezh2或Kdm2a的敲除使肿瘤对抗PD-1治疗敏感(图S1H-K)。值得注意的是,靶向Cbx4(经典多梳抑制复合物的一个组分)的sgRNA在耗竭最显著的sgRNA中排名靠前,突显了其在肿瘤免疫逃逸中的关键作用(图1F)。此外,我们确定了一些因子,其中许多在Hepa1-6或MC38中优先得分。Hepa1-6特异性 hits 包括Sf3b5、Exosc3、Dr1、Exosc9、Eif3k、Dnmt1等。MC38特异性 hits 包括Rbm17、Cul4a、Mettl1、Mettl14、Kat14、Ino80等。这些背景特异性基因也值得进一步研究(图S1E)。
为了进一步研究CBX4在HCC进展中的临床和病理学相关性,我们组建了一个包含108例HCC患者的内部队列(同济队列2,表S2)。排除脱落样本后,共纳入92例配对HCC及癌旁正常组织样本。CBX4在HCC组织中的表达显著高于癌旁正常组织,且在晚期肿瘤中的表达高于早期肿瘤(图1G和图S2)。同时,对多个临床队列(包括50例分期HCC患者(同济队列3,表S2)以及TCGA和CPTAC数据)的综合分析表明,在多种癌症类型中,CBX4在肿瘤组织中的表达均显著高于癌旁非肿瘤组织(图S2G-H)。进一步分析显示,高CBX4表达与更具侵袭性的肿瘤表型密切相关(图1H)。结合免疫组化评分、转录组、蛋白质组数据、免疫细胞浸润和肿瘤纯度的预后评估一致表明,CBX4表达升高与不良临床结局相关(图1G-I和图S2A-M)。值得注意的是,单变量和多变量Cox回归分析均将CBX4确定为独立的危险因素,突显了其在患者风险分层中的潜在应用价值(图S2N-O)。
我们接下来研究了CBX4与ICB疗效之间的关系。临床上,在多种癌症类型(包括肝细胞癌(同济队列4)(图1J-K;表S2)、结直肠癌、黑色素瘤和尿路上皮肿瘤)中,高CBX4表达与免疫治疗耐药相关(图S3A-C)。为了证实这些关联,我们分析了接受新辅助抗PD-1治疗患者的单细胞RNA测序和空间转录组数据(26),结果显示CBX4在肿瘤细胞和免疫抑制性肿瘤相关巨噬细胞亚群中高表达(图1L-N,和图S3 D-I),并且在非应答者的肿瘤组织中显著富集(图1N和图S3F)。接下来,我们使用JASPAR和PROMO进行转录因子结合基序分析,以鉴定Cbx4的潜在调节因子,确定Maz、E2f1、Usf1和Patz1为首选候选因子(图S4A)。siRNA介导的Maz敲低抑制了Hepa1-6细胞中Cbx4的表达,而在相同条件下,E2f1、Usf1和Patz1敲低对Cbx4表达无影响(图S4B-C)。然后,我们利用JASPAR数据库预测了Cbx4启动子区域上的三个Maz结合位点(P1、P2、P3)。Cut&Tag qPCR证实了Maz与Cbx4启动子上P1和P2位点的结合(图S4D-E)。对多个转录组数据集的关联分析显示,MAZ与CBX4表达呈正相关(图S4F-G)。随后,我们收集了免疫治疗后肝细胞癌患者的组织标本。蛋白质印迹分析显示,与应答者相比,免疫治疗非应答者中MAZ和CBX4的水平显著升高(图S4H-I)。此外,CBX4和MAZ的蛋白水平之间存在显著正相关(图S4K)。总之,这些数据表明,通过体内CRISPR筛选鉴定出的CBX4与癌症患者的肿瘤恶性程度和免疫治疗耐药性密切相关。
2.肿瘤细胞中CBX4的缺失诱导由CD8+ T细胞和NK细胞介导的抗肿瘤免疫
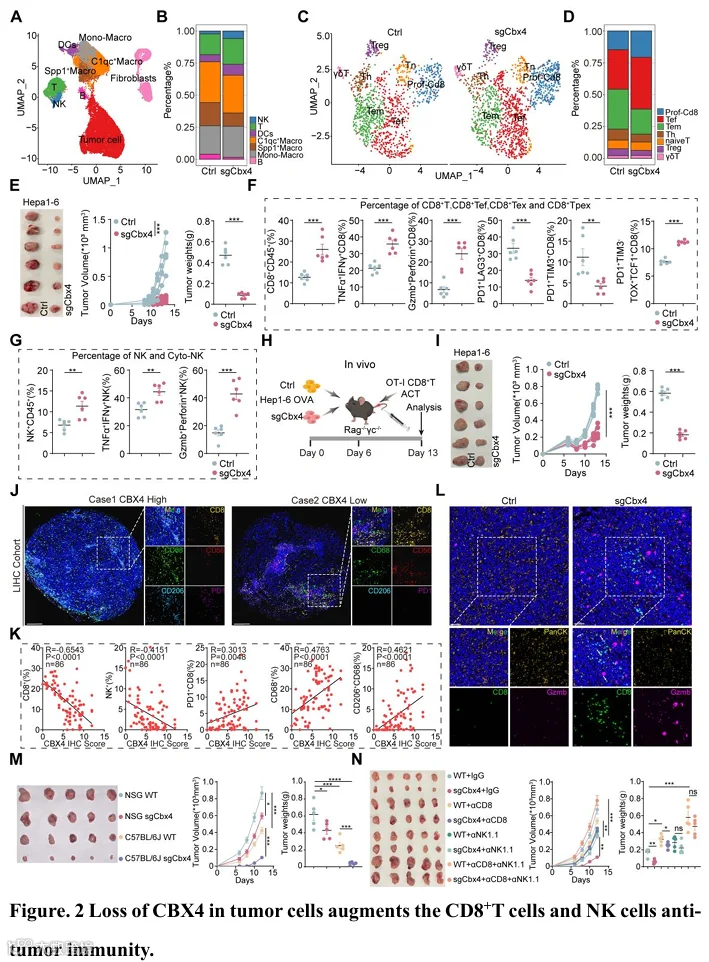
为了全面评估Cbx4对肿瘤微环境的影响,我们对荷Hep1-6对照或Hep1-6 sgCbx4肿瘤的小鼠模型中分离的细胞进行了单细胞RNA测序(图2A)。无监督流形近似和投影(UMAP)聚类鉴定出六种主要细胞类型(图S5A)。在转导sgCbx4的肿瘤中,瘤内CD45+免疫细胞亚群显示T细胞、NK细胞和DC显著增加,同时Spp1+巨噬细胞显著减少(图2B)。为了进一步研究肿瘤细胞中Cbx4缺失对T细胞亚群的影响,我们对T细胞进行了额外的降维分析(图2C)。我们将T细胞定义为7个亚群,包括效应T细胞(Tef)、增殖性CD8+ T细胞、效应记忆T细胞(Tem)、Treg细胞、初始T细胞、辅助T细胞(Th)和γδT细胞(图S5B)。值得注意的是,在Cbx4缺失的肿瘤组织中,具有细胞毒功能的效应T细胞和增殖性CD8+ T细胞的比例显著增加,同时Treg细胞显著减少(图2D)。肿瘤细胞中Cbx4的缺失显著抑制了Hepa1-6和MC38肿瘤的生长(图2E和图S5L)。流式细胞术分析验证了CD8+ T、CD8+ Tef(IFN-γ+ TNF-α+ 和 Perforin+ granzyme B+)、CD8+ Tpex(PD1+ TIM3- TOX+ TCF1+)、NK和cyto-NK(IFN-γ+ TNF-α+ 和 Perforin+ granzyme B+)的比例和数量增加,而Tex细胞的比例减少(图2F-G,图S5C-K,S5M-T和图S6A)。与这些发现一致,在原位HCC模型中也观察到了类似的结果(图S7A-G)。
为了阐明肿瘤细胞中cbx4缺失引发瘤内免疫微环境激活的潜在机制,我们分析了单细胞测序数据集中的肿瘤细胞簇。基因本体论(GO)和基因集富集分析(GSEA)显示,Cbx4缺失诱导了先天免疫通路的激活,包括通过MHC I类分子进行肽抗原的抗原加工和呈递、对干扰素-β的反应以及先天免疫反应的正向调节(图S8A-B)。接下来,我们分析了sgCbx4肿瘤细胞中与T细胞刺激相关的基因表达。肿瘤细胞表现出抗原呈递机制(B2m、H2-D1)的表达上调,这对T细胞存活和效应分化至关重要(27,28)。此外,Cxcl10(一种对T和NK细胞募集至关重要的趋化因子,与良好的免疫治疗反应相关(29,30))也高水平表达(图S8C)。为了进一步阐明Cbx4对肿瘤微环境的影响,我们进行了一系列体外和体内实验。在共培养实验中,当与表达OVA的Cbx4缺陷型肿瘤细胞(而非NC肿瘤细胞)孵育时,OVA特异性CD8+ T细胞显示出增殖和效应细胞因子产生的显著增加(图S8D-F)。此外,我们将表达OVA的Hepa1-6细胞皮下植入T、B和NK细胞缺陷的Rag-/-γc-/-小鼠中,并通过过继转移OVA特异性CD8+ T细胞模拟CD8+ T细胞介导的免疫压力(图2H)。Cbx4缺陷型肿瘤显著增强了过继转移的T细胞抑制肿瘤生长的功效(图2I)。在将初始NK细胞与Hepa1-6肿瘤细胞共培养时也观察到了类似的结果(图S8G)。我们证实,肿瘤细胞中Cbx4的缺失也使NK细胞能够有效分化为“细胞毒性”表型(IFN-γ+ TNF-α+ 和 Perforin+ granzyme B+)(图S8H)。此外,我们构建了Vector-tagBFP和Cbx4-Sh-tagGFP肿瘤细胞,并以1:1的比例混合进行肿瘤排斥实验(图S8I)。Cbx4-Sh-tagGFP肿瘤细胞更有可能被清除,并且PD-1阻断增强了它们的脆弱性(图S8J),这与临床免疫治疗队列和体内CRISPR筛选中的观察结果一致,即Cbx4低表达预示着更好的总生存期。此外,多重免疫组化显示,小鼠中肿瘤细胞Cbx4缺陷通过增加肿瘤细胞与CD8+ T细胞之间的接触面积,增强了CD8+ T细胞浸润和细胞毒性(图2L)。一致地,来自同济队列2的多重免疫组化(图2J)显示,CBX4表达与CD8+ T细胞和NK细胞的丰度呈显著临床负相关(图2K),而与耗竭T细胞和M2巨噬细胞的存在呈显著正相关(图2K)。
鉴于肿瘤中Cbx4缺失导致CD8+ T细胞和NK细胞的变化,我们首先阐明了sgCbx4对肿瘤生长的抑制作用是否依赖于免疫。我们的数据显示,在NSG小鼠中,靶向Cbx4仅轻微抑制肿瘤生长(图2M)。与先前研究一致,Cbx4缺失可以抑制肿瘤细胞的增殖(31)。然而,在免疫健全的C57BL/6J小鼠中,靶向肿瘤细胞中的Cbx4显著抑制了肿瘤生长。这些结果表明,靶向肿瘤细胞中Cbx4的抗肿瘤功效更依赖于免疫细胞的功能(图2M)。为了探索靶向Cbx4抑制肿瘤进展过程中需要哪些特定的免疫细胞亚群,使用耗竭抗体选择性地清除CD8+ T细胞、NK细胞或CD8+ T/NK细胞(图S8K)。我们的结果表明,清除CD8+ T细胞和NK细胞消除了Cbx4缺失对肿瘤进展的影响(图2N)。这些发现表明,靶向Cbx4依赖于CD8+ T细胞和NK细胞抑制肿瘤生长,这促使我们集中研究Cbx4促进肿瘤细胞抵抗CD8+ T细胞和NK细胞依赖性细胞毒性的潜在机制。
3.巨噬细胞中CBX4的缺失通过CD8+ T细胞和NK细胞诱导抗肿瘤免疫
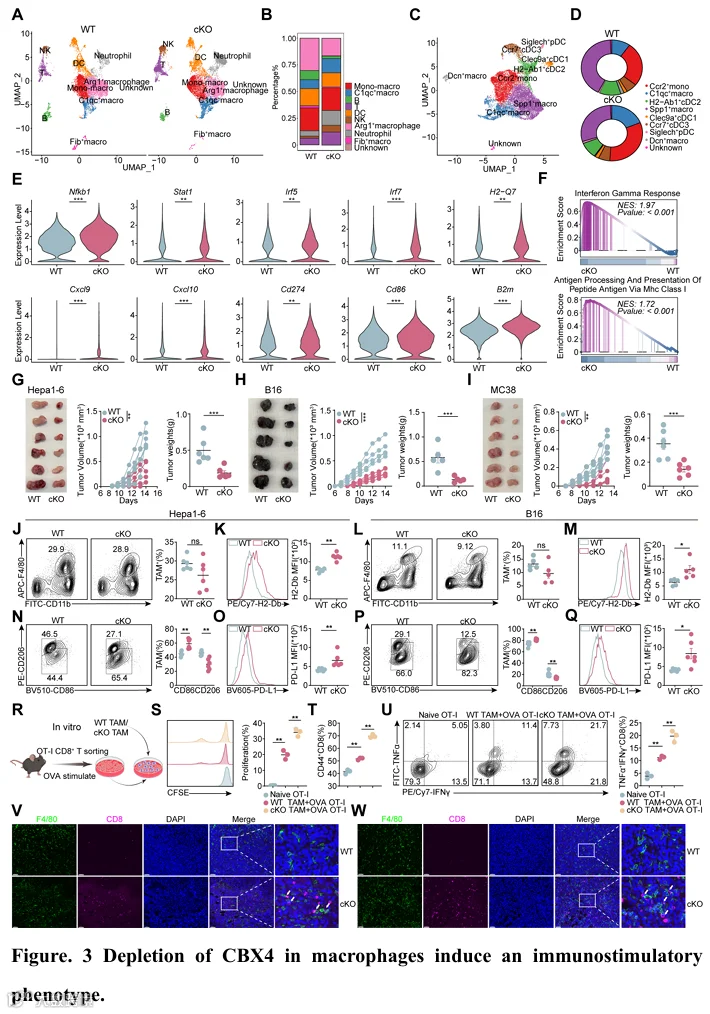
基于接受新辅助抗PD-1治疗患者的单细胞RNA测序和空间转录组学,我们观察到CBX4在非应答者肿瘤组织中的免疫抑制性TAM亚群中高表达,表明巨噬细胞中的CBX4也可能在免疫逃逸中发挥关键作用(图1M和图S3F)。免疫荧光染色显示,肝细胞癌组织中CBX4和巨噬细胞显著共定位(图S9A)。此外,CBX4的表达水平与CD68(巨噬细胞标志物)呈高度正相关,且高CBX4和CD68表达与不良临床结局相关(图S9A-C)。因此,我们构建了LysM Cre Cbx4 fl/fl 髓系细胞条件性敲除小鼠。为了全面评估髓系细胞中Cbx4缺失对肿瘤微环境的影响,我们富集了CD45+免疫细胞,对从野生型和LysM Cre Cbx4 fl/fl 小鼠的Hep1-6肿瘤组织中分离的细胞进行了单细胞RNA测序(图3A)。我们鉴定出10个不同的细胞簇,包括T细胞、NK细胞、B细胞、DC、C1qc+巨噬细胞、Arg1+巨噬细胞、成纤维细胞样巨噬细胞、单核细胞和一个未知细胞亚群(图S10A)。跨条件的比较簇动力学分析显示,在LysM Cre Cbx4 fl/fl 小鼠的肿瘤中,T细胞、NK细胞和C1qc+巨噬细胞(被认为是M1样亚群)(32)显著增加,同时Arg1+巨噬细胞(被认为是M2样免疫抑制亚群)(32)显著减少(图3B)。为了进一步研究髓系细胞中Cbx4缺失的影响,我们对髓系细胞进行了额外的降维分析(图3C)。我们将髓系细胞定义为9个亚群,包括C1qc+巨噬细胞、Spp1+巨噬细胞、Ccr2+巨噬细胞、Dcn+巨噬细胞、Cle9a+ cDC1、H2-Ab1+ cDC2、Ccr7+ cDC3、Siglech+ pDC和一个未知亚群(图S10B)。值得注意的是,M1样C1qc+巨噬细胞和Ccr2+巨噬细胞的比例显著增加,同时免疫抑制性Spp1+巨噬细胞显著减少(图3D)。然而,Cle9a+ cDC1、H2-Ab1+ cDC2、Ccr7+ cDC3、Siglech+ pDC的比例仅显示轻微变化(图3D),并且DC中Cbx4的表达水平显著较低(图S3H),这表明Cbx4的缺失主要通过巨噬细胞相关亚群发挥其功能。值得注意的是,基因差异分析和GSEA显示,巨噬细胞中Cbx4的缺失促进了先天免疫通路的激活(图3E-F)。为了验证我们的单细胞发现,我们对HCC、黑色素瘤和结直肠癌小鼠模型进行了流式细胞术。巨噬细胞中Cbx4的缺失显著抑制了肿瘤生长(图3G-I)。与单细胞发现一致,来自HCC和黑色素瘤的LysM Cre Cbx4 fl/fl 组的流式细胞术结果显示,总肿瘤相关巨噬细胞的比例没有显著变化(图3J和L)。然而,LysM Cre Cbx4 fl/fl 组中CD86+ TAM的比例显著更高,同时免疫抑制性CD206+巨噬细胞显著减少(图3N和P)。同时,MHC I类分子和PD-L1的表达显著增加(图3K、M、O和Q)。为了阐明巨噬细胞的抗原呈递能力,我们将初始OT-I CD8+ T细胞与负载ova257-264肽的野生型或LysM Cre Cbx4 fl/fl BMDM共培养。一致地,共培养实验和流式细胞术显示,Cbx4缺失的TAM表现出增殖和效应细胞因子产生的显著增加(图3R-U)。此外,来自Hep1-6和B16荷瘤组织的多重免疫组化显示,巨噬细胞中Cbx4缺陷通过增加巨噬细胞与CD8+ T细胞之间的接触面积,增强了CD8+ T细胞浸润,表明在Cbx4缺失下巨噬细胞与CD8+ T细胞之间存在更多的细胞间相互作用和通讯(图3V-W)。
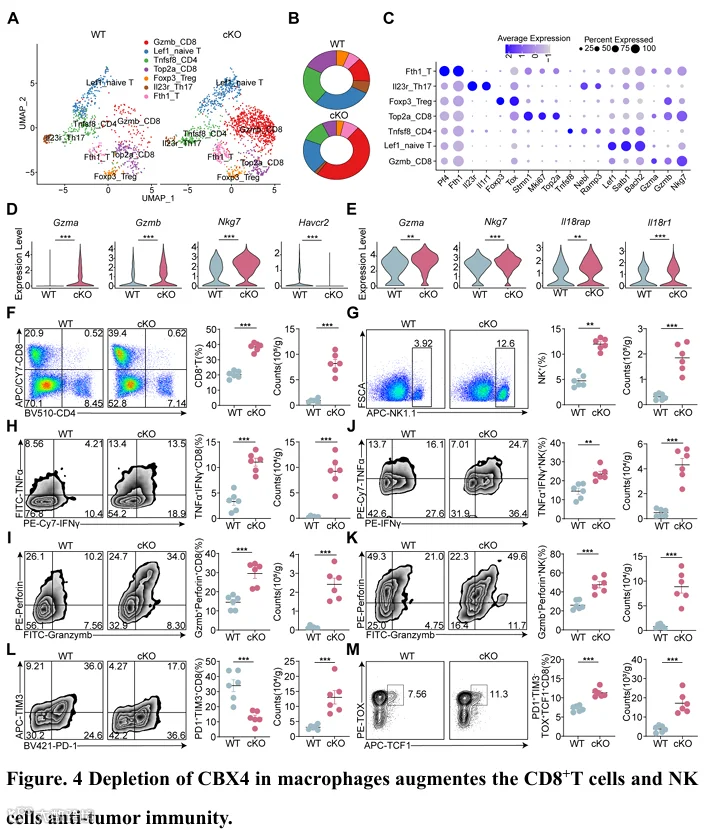
为了进一步研究巨噬细胞中Cbx4缺陷对T细胞亚群的影响,我们对T细胞进行了额外的降维分析(图4A)。我们将T细胞定义为7个亚群,包括Gzmb+ CD8+ T细胞(Tef)、Top2+ CD8+ T细胞(增殖性CD8+ T细胞)、Tnfsf8+ T细胞、Treg细胞、Left+ T细胞(初始T细胞)、Il23r+ Th17细胞和Fth1+ T细胞(图4C)。值得注意的是,在LysM Cre Cbx4 fl/fl 小鼠的肿瘤中,具有细胞毒功能的效应CD8+ T细胞的比例和数量显著增加,同时Treg细胞显著减少(图4B)。T细胞和NK细胞中的差异表达基因显示,巨噬细胞中Cbx4的缺失重塑了炎性肿瘤微环境(图4D-E)。来自HCC和黑色素瘤的流式细胞术结果验证了CD8+ T(图4F和图S11A)、CD8+ Tef(IFN-γ+ TNF-α+ 和 Perforin+ granzyme B+)(图4H-I和图S11C-D)、CD8+ Tpex(PD1+ TIM3- TOX+ TCF1+)(图4M)、NK(图4G和图S11B)和cyto-NK(图4J-K和图S11E-F)的比例和数量增加,而Tex细胞(PD1+ TIM3+ 和 PD1+ LAG3+)的比例减少(图4L和图S11G-H)。与这些发现一致,在原位HCC模型中也观察到了类似的结果(图S12A-G)。此外,多重免疫组化显示,巨噬细胞中Cbx4缺陷增强了CD8+ T细胞和NK细胞的浸润和细胞毒性(图S11I)。鉴于巨噬细胞中Cbx4缺失导致CD8+ T细胞和NK细胞的变化,我们通过选择性清除CD8+ T细胞、NK细胞或两者来进一步阐明这些特定免疫细胞亚群的作用(图S11J)。我们的结果表明,清除CD8+ T细胞和NK细胞消除了巨噬细胞中Cbx4缺失对肿瘤进展的影响(图S11J-K)。这些发现表明,巨噬细胞中Cbx4缺失以CD8+ T细胞和NK细胞依赖的方式促进抗肿瘤免疫,这可能与肿瘤细胞中Cbx4缺陷的机制相似。
4.CBX4缺失通过激活RIG-I-干扰素信号触发抗病毒免疫应答
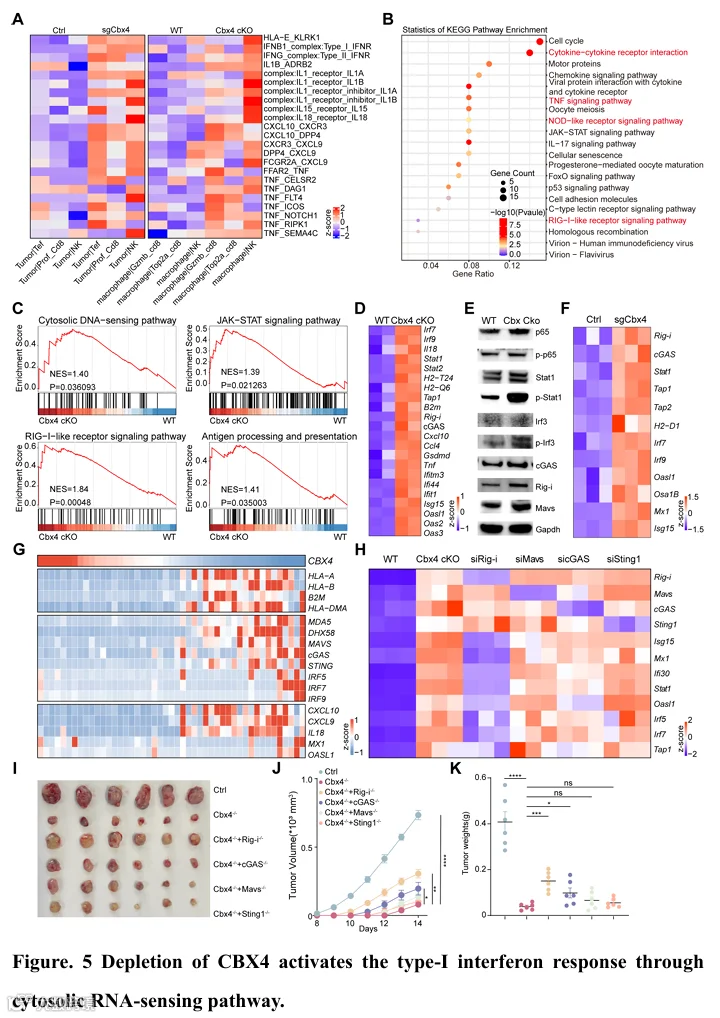
T细胞和NK细胞在其效应功能中并非自主的。靶向再激活T细胞和NK细胞需要协调的细胞间相互作用和通讯,包括趋化因子的分泌、干扰素相关通路和抗原呈递通路的激活(6,33-35)。鉴于肿瘤细胞和巨噬细胞中的Cbx4缺陷均触发CD8+ T和NK细胞介导的抗肿瘤免疫,我们利用CellPhoneDB基于scRNA-seq数据推断肿瘤细胞/巨噬细胞与T细胞/NK细胞之间的相互作用(图5A)。值得注意的是,我们观察到Cbx4缺陷的肿瘤细胞/巨噬细胞与T细胞/NK细胞之间的相互作用显著增加,包括炎症细胞因子(如TNFα、IFNβ、IFNγ和白介素-1家族成员IL1b、IL15和IL18(对T细胞和NK细胞存活及效应分化至关重要)(35-37))、趋化因子(如Cxcl9和Cxcl10(对T和NK细胞募集至关重要)(15,29,30))以及HLA-E-NKG2D轴(对激活T细胞和NK细胞的细胞毒功能至关重要)(38,39)的相互作用(图5A)。随后,我们对Cbx4缺陷的肿瘤细胞/巨噬细胞与对照进行了KEGG和GSEA分析,以查询标志性通路数据库。分析显示,肿瘤细胞和巨噬细胞中Cbx4缺失均导致多个免疫相关通路的上调,如I型干扰素反应、RIG-I样受体信号和胞质DNA感知通路、抗原加工和呈递通路等(图5B-C和图S13A)。一致地,肿瘤细胞和巨噬细胞中Cbx4缺陷导致RIG-I、MAVS、cGAS以及磷酸化IRF3、IRF7、STAT1和Nfkb的mRNA和蛋白水平升高(图5D-F)。临床上,来自HCC患者(同济队列)、黑色素瘤和结直肠癌肿瘤组织的差异表达基因进一步揭示,CBX4表达与参与抗原呈递(MHC I相关)、先天免疫和T细胞活化的基因呈负相关(图5G和图S13B-C)。一致地,多重细胞因子检测显示,在Cbx4缺陷的肿瘤细胞/巨噬细胞荷瘤小鼠中,炎症细胞因子广泛上调(图S13D-E)。
我们接下来通过敲低不同的dsDNA和dsRNA传感器,评估了先天免疫受体通路在Cbx4缺失诱导的IFN激活中的作用。有趣的是,cGAS的缺失仅轻微降低了巨噬细胞中Cbx4缺失诱导的IFN信号,表明cGAS/Sting1通路似乎不是Cbx4缺失诱导IFN信号的关键介质(图5H)。相反,RIG-I的缺失完全逆转了Cbx4缺失诱导的ISG和炎症基因表达,表明RIG-I是关键介质(图5H)。为了确定Cbx4是否主要通过RIG-I样受体信号通路影响肿瘤进展和免疫微环境重塑,我们构建了稳定的Cbx4/Rig-i双敲除、Cbx4/cGAS双敲除、Cbx4/Mavs双敲除和Cbx4/Sting1双敲除的Hepa1-6细胞系(图5I)。荷瘤小鼠模型数据和多重细胞因子检测显示,Rig-i缺失削弱了与Cbx4缺陷相关的肿瘤抑制和免疫调节效应(图5I-K和图S13F)。总之,这些发现表明,肿瘤细胞和巨噬细胞中Cbx4的缺陷介导了一种共同的机制,即通过RIG-I受体信号通路重塑肿瘤微环境。
5.CBX4抑制H3K9me3和H3K27me3标记的内源逆转录转座子
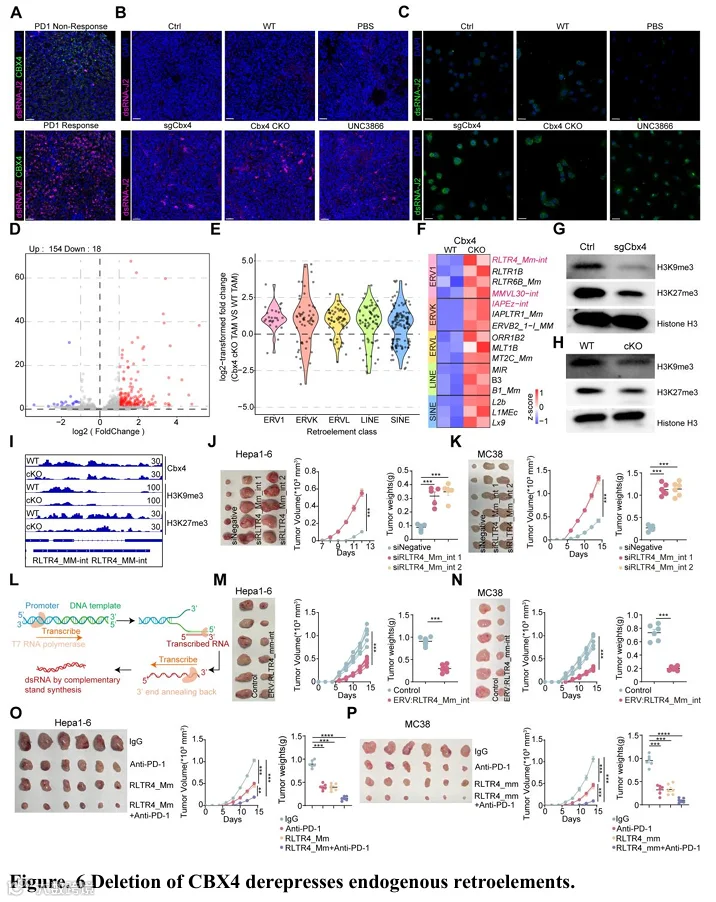
RIG-I受体信号通路可被内源双链RNA激活,包括错误剪接的转录本和内源逆转录病毒(ERV),它们是胞质dsRNA的主要贡献者(40,41)。然而,在Cbx4缺陷的肿瘤细胞或巨噬细胞中,剪接体相关通路没有富集(图S8A和图5B)。我们推测Cbx4缺陷激活了ERV和内源核酸感知通路。使用dsRNA特异性抗体(J2)染色,我们在人样本中检测到,与高CBX4表达的非应答者相比,低CBX4表达的抗PD-1应答者中dsRNA水平更高(图6A)。此外,体外和体内实验均证实,靶向Cbx4诱导了细胞质和肿瘤组织中dsRNA的积累(图6B-C)。RNA-seq分析显示,来自逆转录转座子的双向转录本增加,包括含有长末端重复序列的ERV和非LTR元件,例如RLTR4-Mm-int,这是在TAM中Cbx4缺失诱导的最主要的ERV(图6D-F)。在Hepa1-6肿瘤细胞中也观察到了类似的结果(图S14A)。调节ERV表达的基本表观遗传机制包括DNA甲基化和组蛋白修饰(42)。然而,MethylFlash全球DNA甲基化(5-mC)ELISA检测显示,巨噬细胞和肿瘤细胞中Cbx4缺失并未显著改变全局DNA甲基化水平,表明Cbx4缺失引起的ERV表达激活可能是由组蛋白甲基化修饰诱导的(图S14B-C)。逆转录转座子的沉默主要由抑制性H3K9me3和H3K27me3异染色质控制(43-45)。最近的研究表明,作为PRC1复合物一部分的Cbx4,可与组蛋白H3在“Lys-9”位点的三甲基化(H3K9me3)和H3在“Lys-27”位点的三甲基化(H3K27me3)结合(46),表明Cbx4可能通过染色质重塑和组蛋白修饰来调节ERV表达。接下来,我们证明了TAM和肿瘤细胞中Cbx4缺陷降低了H3K9me3水平和H3K27me3水平(图6G-H)。Cbx4的缺失导致H3K9me3和H3K27me3抑制性标记在RLTR4-MM-int位点的结合减少(图6I和图S14E)。
在巨噬细胞中敲低最主要的Cbx4缺失诱导的ERV——RLTR4-Mm-int,降低了I型干扰素反应,而不影响其他ERV的表达(图S14D和F)。此外,为了确定Cbx4是否主要通过激活RLTR4-Mm-int影响肿瘤进展和免疫微环境重塑,我们构建了稳定的Cbx4/RLTR4-Mm-int双敲低的Hepa1-6/MC38细胞系(图6J-K)。我们的发现表明,RLTR4-Mm-int敲低恢复了与Cbx4缺陷相关的肿瘤抑制和免疫调节效应(图6J-K)。为了更好地确定RLTR4-Mm-int的潜在治疗价值,我们设计了用于体内治疗的ERV:RLTR4-Mm-int(图6L)。瘤内注射ERV:RLTR4_Mm_int显著抑制了Hep1-6和MC38肿瘤的生长(图6M-N)。流式细胞术显示,ERV:RLTR4_Mm_int增加了CD8⁺ T、CD8⁺ Tef(IFN-γ⁺ TNF-α⁺ 和 Perforin⁺ granzyme B⁺)、CD8⁺ Tpex(PD1⁺ TIM3⁻ TOX⁺ TCF1⁺)、CD8⁺ Tprof(Ki67⁺)、早期活化CD8⁺ T(CD69⁺)、NK和cyto-NK(IFN-γ⁺ TNF-α⁺ 和 Perforin⁺ granzyme B⁺)的比例,而Tex细胞(PD1⁺ TIM3⁺ 和 PD1⁺ LAG3⁺)的比例减少(图S15A-B)。体外和体内实验表明,ERV:RLTR4_Mm_int诱导炎症因子的表达和释放,从而重塑免疫抑制性肿瘤微环境(图S15C-F)。然后,我们在免疫健全小鼠的HCC和CRC模型中引入了ERV:RLTR4_Mm_int与PD-1阻断的单药或联合治疗(图6O-P)。正如假设的那样,其与免疫检查点阻断的联合进一步增强了治疗效力,产生了强效且持久的抗肿瘤反应(图6O-P)。
为了探索CBX4与已知ERV表达表观遗传调节因子之间的潜在相互作用,我们使用抗CBX4抗体进行了免疫沉淀结合质谱分析(IP-MS)。我们的结果表明,CBX4与表观遗传调节因子EZH2(主要靶向H3K27me3)和SETDB1(主要靶向H3K9me3)特异性相互作用(47,48),表明CBX4可能通过与这些因子形成多蛋白复合物来抑制ERV表达。相反,我们没有观察到CBX4与EP300或KDM5B之间的相互作用(图S16A-C)。为了评估组蛋白甲基化的药理调节是否能逆转CBX4介导的免疫抑制,我们使用针对组蛋白标记H3K9me3和H3K27me3的特异性抑制剂进行了挽救实验,这些标记受CBX4调节。我们选择了SETDB1特异性抑制剂(SETDB1-TTD-IN-1)和EZH2特异性抑制剂(GSK126)在随后的挽救实验中药理学抑制H3K9me3和H3K27me3水平。然后,我们构建了稳定过表达Cbx4的肿瘤细胞系,并进行了皮下肿瘤植入实验。正如预期的那样,Cbx4过表达显著减少了肿瘤浸润的CD8+ T细胞和NK细胞的比例。重要的是,用H3K9me3或H3K27me3抑制剂治疗有效地破坏了这种免疫抑制微环境,恢复了免疫细胞浸润并抑制了肿瘤进展(图S16D-M)。这些数据表明,靶向组蛋白甲基化可以在功能上逆转CBX4诱导的免疫逃逸。
为了更好地增强临床相关性,我们还分析了抗PD-1治疗前采集的黑色素瘤预处理肿瘤样本的RNA-seq数据集(49)。我们发现CBX4表达与ERV(如ERVmap_2550、ERVmap_2192、ERVmap_705等)呈负相关,这些ERV在对抗PD-1治疗有完全缓解的患者中表达显著高于疾病进展患者(图S17A-C)。总之,我们证明了肿瘤细胞和巨噬细胞中Cbx4的缺陷介导了一种共同的机制,即通过去抑制H3K9me3和H3K27me3标记的内源逆转录转座子RLTR4-MM-int来重塑肿瘤微环境。ERV:RLTR4_Mm_int可能作为增强抗肿瘤免疫和癌症免疫治疗的治疗靶点。
6.靶向CBX4诱导抗肿瘤免疫并增强PD-1阻断的体内抗肿瘤效果
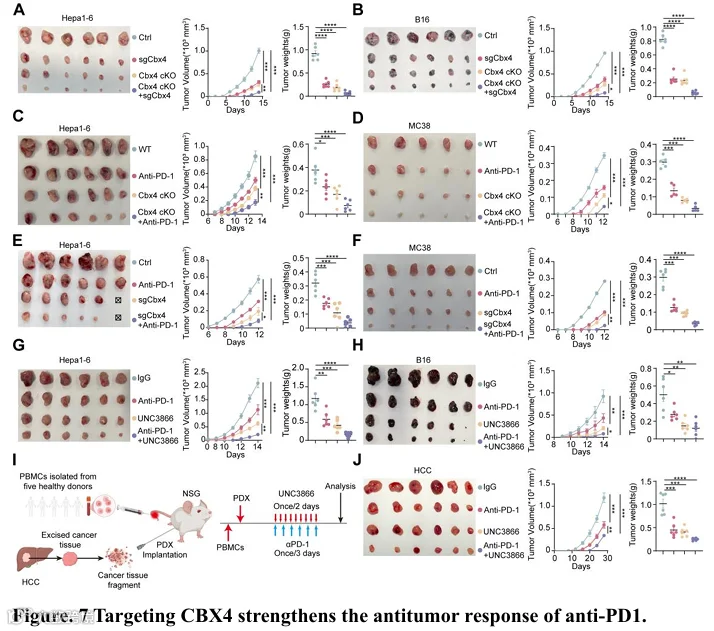
为了更好地理解同时缺失两个区室中的Cbx4将如何影响肿瘤进展和免疫反应,我们使用Hepa1-6和B16荷瘤小鼠模型,比较了单独缺失肿瘤细胞和巨噬细胞中Cbx4以及联合缺失Cbx4的效果。联合缺失Cbx4显著抑制了肿瘤生长,并诱导了更多的效应T细胞和NK细胞(图7A-B和图S18A-F)。接下来,使用Hepa1-6和mc38荷瘤小鼠模型,我们证明巨噬细胞(图7C-D)或肿瘤细胞(图7E-F)中CBX4缺失均能增强抗PD-1免疫治疗的疗效。一致地,我们观察到抗PD-1与巨噬细胞中CBX4缺失的联合治疗并未改变总肿瘤相关巨噬细胞的比例(图S19A),但增加了MHC I类分子的表达(图S19B)。然而,联合治疗组中CD86+ TAM的比例显著更高,同时免疫抑制性CD206+巨噬细胞显著减少(图S19C-D)。重要的是,细胞毒性CD8+ T细胞、NK细胞和细胞毒性TNFα+ IFN-γ+ CD8+ T细胞的比例显著增加(图S19E-G),联合治疗进一步增强了这一效应。为了评估靶向CBX4的治疗价值,我们用CBX4抑制剂UNC3866、抗PD-1抗体或其联合治疗Hepa1-6和B16荷瘤小鼠,并监测肿瘤生长。我们注意到,在Hepa1-6和B16荷瘤小鼠模型中,CBX4抑制与抗PD-1抗体的联合使用显著减缓了肿瘤生长(图7G-H)。一致地,我们观察到总肿瘤相关巨噬细胞的比例没有显著变化(图S19H)。然而,联合治疗组中CD86+ TAM的比例显著更高,同时免疫抑制性CD206+巨噬细胞显著减少(图S19J-K)。同时,具有抗原呈递能力的MHC I类分子的表达显著增加(图S19I)。重要的是,UNC3866单药治疗后CD8+ T细胞、NK细胞和细胞毒性TNFα+ IFN-γ+ CD8+ T细胞的比例显著增加,联合治疗进一步增强了这一效应(图S19L-N)。UNC3866在Rag-/-γc-/-小鼠中仅轻微抑制肿瘤生长。然而,在免疫健全的C57BL/6J小鼠中,UNC3866显著抑制了肿瘤生长,并增加了CD8+ T细胞和NK细胞的比例(图S19Q-R)。这些结果表明,UNC3866通过免疫介导的方式限制肿瘤生长,更依赖于免疫细胞的功能。为了更好地模拟对免疫治疗的临床反应,我们使用来自具有不同CBX4表达和免疫治疗结果的个体的新鲜切除HCC标本,在人性化NSG小鼠中建立了患者来源异种移植(PDX)模型。与对照组相比,UNC3866单药治疗显著抑制了肿瘤生长并减少了肿瘤负荷。正如假设的那样,其与免疫检查点阻断的联合进一步增强了治疗效力,产生了强效且持久的抗肿瘤反应(图7I-J)。总之,这些发现表明,CBX4是一个有前景的治疗靶点,能使肿瘤对免疫检查点阻断敏感,为增强抗肿瘤免疫提供了一种引人注目的联合治疗策略。这些结果突显了表观遗传-免疫共靶向作为一种增强癌症免疫治疗的有前景且临床可行的策略。
更多结果和补充图表:doi: 10.1172/JCI200564
长按二维码关注我们,用最短的时间和最高的效率学习更多数据分析方法!
扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:
https://cnsknowall.com
CNSknowall:24年最新问世的遥遥领先的科研数据(0代码生信+统计学)分析平台,同时含有机制图模块+汉化版Pubmed融合Deepseek高效筛选目标文献+SCI文献例句/语料检索模块+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动批阅!