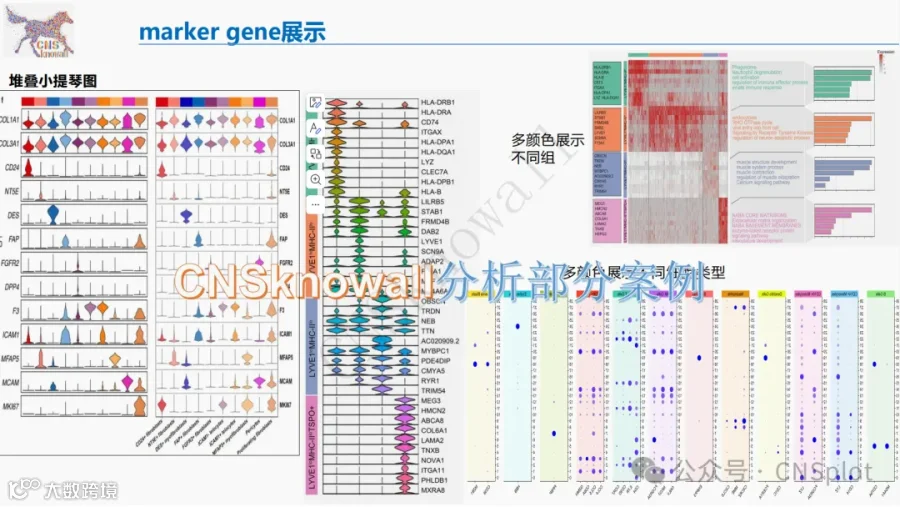
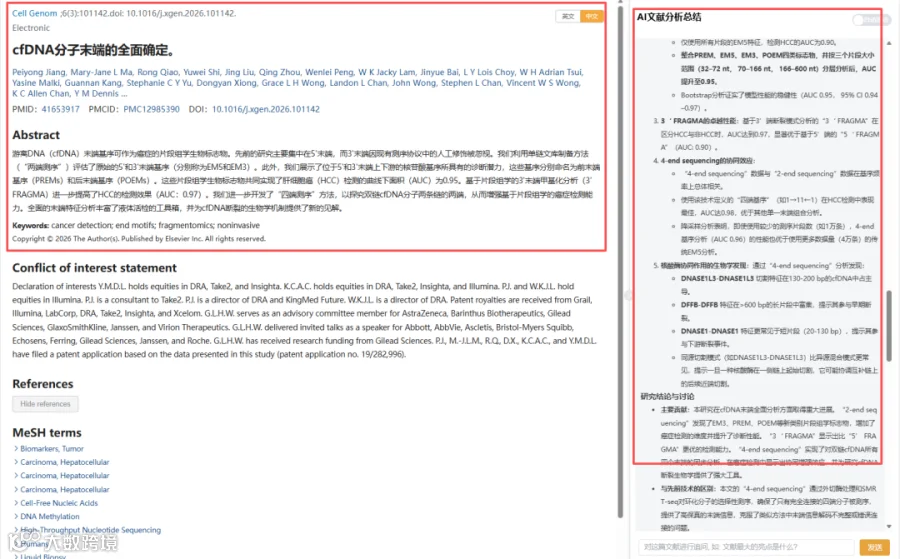
传统 cfDNA 研究多聚焦 5′ 端,而 3′ 端因建库过程被破坏长期被忽视。本研究通过单链建库(2‑端测序)首次同时捕获 cfDNA 的 5′ 与 3′ 天然末端,并进一步开发茎环接头‑环化测序(4‑端测序),实现双链 DNA 四个末端的全息解析。研究定义了 PREM、EM5、EM3、POEM 四类末端基序,结合片段大小分层分析,肝癌检测 AUC 达 0.95;基于 3′ 末端的 FRAGMA 分析 AUC 提升至 0.97;而整合四个末端的 4‑端测序更将 AUC 推至 0.98。该工作将 cfDNA 片段组学从“单端”推向“全景”,为液体活检提供了全新维度的生物学与临床信息。
今天给大家解读一篇3月发表在《Cell Genomics》上的题目为“Holistic determination of ends of cfDNA molecules.”的文章。本文针对传统cfDNA片段组学研究主要关注5‘端而忽视3’端的局限,开发了两种新型测序策略。通过“2-end sequencing”,本研究全面刻画了cfDNA的EM5、EM3、PREM和POEM四类末端基序的特征,发现EM5/POEM、PREM/EM3分别具有高度相似性,且这些基序主要与核酸酶DNASE1L3相关。在HCC检测中,整合这四类标志物并分层大小分析,可将诊断AUC提升至0.95。基于3‘端开发的“3’ FRAGMA”方法进一步将AUC提升至0.97。随后,本研究首创“4-end sequencing”,实现了对双链cfDNA四个末端的同步分析,其标志物(1→11←1)在HCC检测中达到AUC 0.98,且所需数据量少于传统EM5分析。此外,利用该技术揭示了不同核酸酶在长短cfDNA片段断裂中的动态与协同参与。研究认为,对cfDNA末端的全面分析为液体活检增添了强大的新工具,并深化了对cfDNA产生生物学的理解。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!


团队成员合影(位于上海陆家嘴中心,可随时预约参观)
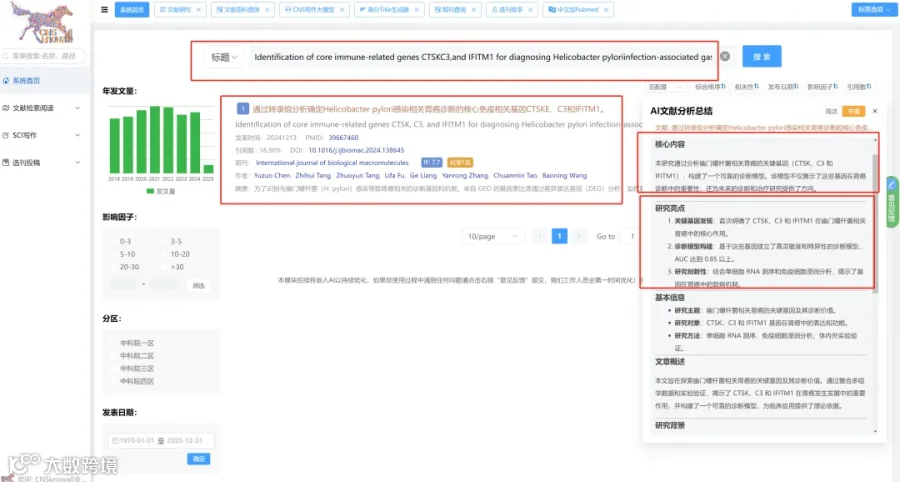
题目:《cfDNA分子末端的全面确定》Holistic determination of ends of cfDNA molecules
发表期刊:Cell Genomics
影响因子:9
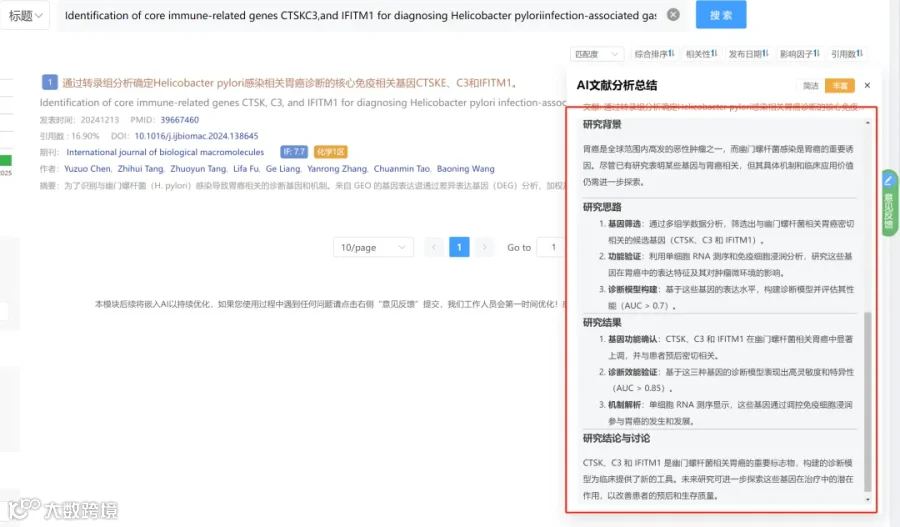
研究背景:
-
cfDNA片段组学是液体活检中快速发展的领域。已有研究聚焦于片段大小、偏好末端坐标、5‘端基序(如4-mer)、核小体模式等。
-
既往片段组学研究主要集中于5‘端,这是因为广泛使用的双链DNA(dsDNA)建库方法包含末端修复步骤,会移除或改变3’端的内在特征,使其诊断价值未被探索。
-
单链DNA(ssDNA)建库方法通过变性后直接连接测序接头,可以保留天然5‘和3’端。然而,基于该方法的前期研究并未探讨天然3‘端的诊断相关性。
-
核酸酶(如DNASE1L3)已被证实参与cfDNA的断裂过程,并与某些末端基序(如“CCCA”)的变化相关。
CNSknowall 平台 Pubmed+AI 快速提炼全文要点
研究思路:
- 第一步(2-end sequencing)
采用ssDNA建库技术(Illumina平台),在不进行末端修复的情况下,直接测定单链cfDNA的天然5‘和3’端基序(EM5和EM3),并根据参考基因组推断其上游的PREM和下游的POEM。此策略用于发现和验证包括3‘端在内的新型片段组学标志物在癌症检测中的价值。
- 第二步(3’ FRAGMA)
将先前发表的基于5‘端断裂模式推测甲基化状态的方法(5’ FRAGMA)拓展至3‘端,开发“3’ FRAGMA”,并评估其诊断性能。
- 第三步(4-end sequencing)
为解决“2-end sequencing”无法追溯同一双链分子两端信息的问题,开发新方法。将带有随机单链突出端的定制干环适配体直接连接到双链cfDNA上,通过外切酶处理和PacBio环状一致性测序,确保只分析成功在四个末端都完成连接并环化的分子,从而实现对单个双链cfDNA分子全部四个末端的同步解码。
- 生物学探究
利用“4-end sequencing”技术,通过分析同一侧5‘和3’端基序的组合,探究不同核酸酶(DNASE1L3、DNASE1、DFFB)在产生不同大小cfDNA片段过程中的动态和协调作用。
研究亮点:
- 单链DNA(ssDNA)测序文库制备能够对cfDNA进行3‘端片段组学分析
突破了传统双链建库因末端修复而丢失3’端信息的限制。
- 干环适配体介导的双链DNA(dsDNA)测序能够对全部四个末端进行整体分析
实现了对单个双链cfDNA分子所有末端的同步测定。
- 开发了一套包含5’端、3‘端、前端(Pre-)和后端(Post-)的片段组学标志物体系
- 对末端的整体分析提升了癌症检测性能,并深化了对cfDNA生物学的理解。
研究结果:
- 新型末端基序的特征
在健康人对照中,PREM和EM3的基序模式高度相似(r=0.91),EM5和POEM高度相似(r=0.94)。Top基序分析表明,这些基序在Dnase1l3敲除小鼠中显著下调,提示其主要由DNASE1L3产生。
- HCC检测性能提升
-
与健康对照相比,HCC患者的PREM、EM5、EM3和POEM均存在大量差异表达的4-mer基序。
-
仅使用所有片段的EM5特征,检测HCC的AUC为0.90。
- 整合PREM、EM5、EM3、POEM四类标志物,并按三个片段大小范围(32–72 nt, 70–166 nt, 166–600 nt)分层分析后,AUC提升至0.95
-
Bootstrap分析证实了模型性能的稳健性(AUC 0.95, 95% CI 0.94–0.97)。
- 3‘ FRAGMA的卓越性能
基于3’端断裂模式分析的“3‘ FRAGMA”在区分HCC与非HCC时,AUC达到0.97,显著优于基于5’端的“5‘ FRAGMA”(AUC: 0.90)。
- 4-end sequencing的协同效应
-
“4-end sequencing”数据与“2-end sequencing”数据在基序频率上总体相关。
-
使用该技术定义的“四端基序”(如1→11←1)在HCC检测中表现最佳,AUC达0.98,优于其他单一末端组合分析。
-
降采样分析表明,即使使用较少的测序片段数(如1万条),4-end基序分析(AUC 0.96)的性能也优于使用更多数据量(4万条)的传统EM5分析。
- 核酸酶协同作用的生物学发现
通过“4-end sequencing”分析发现:
- DNASE1L3-DNASE1L3
切割特征在130-200 bp的cfDNA中占主导。
- DFFB-DFFB
特征在>600 bp的长片段中富集,提示其参与早期断裂。
- DNASE1-DNASE1
特征更常见于短片段(20-130 bp),提示其参与下游断裂事件。
-
同源切割模式(如DNASE1L3-DNASE1L3)比异源混合模式更常见,提示一旦一种核酸酶在一侧链上起始切割,它可能协调互补链上的后续近端切割。
研究总结:
- 主要贡献
本研究在cfDNA末端全面分析方面取得重大进展。“2-end sequencing”发现了EM3、PREM、POEM等新类别片段组学标志物,增加了癌症检测的维度并提升了诊断性能。“3‘ FRAGMA”显示出比“5’ FRAGMA”更优的检测能力。“4-end sequencing”实现了对双链cfDNA所有四个末端的同步分析,在癌症检测中显示出协同增强效应,并为研究cfDNA断裂生物学提供了强大工具。
- 与先前技术的区别
本文的“4-end sequencing”通过外切酶处理和SMRT-seq对环化分子的选择性测序,确保了只有完全连接的四端分子被测序,提供了高保真的末端信息,克服了类似方法中末端信息解码不完整或错误连接的问题。
- 意义与展望
对cfDNA末端的全面分析为液体活检增添了强大的新武器,并有助于阐明cfDNA断裂的生物学机制。这些技术发展和新发现的标志物,未来可与其他cfDNA特征(大小、拷贝数、突变、甲基化等)整合,进一步提升液体活检在癌症及其他领域(如产前检测、移植监测)的性能。当前研究的局限性包括临床样本量相对较小、PacBio平台通量有限等,未来通过扩大样本量、提升测序通量以及将方法适配至Illumina等平台,有望推动其更广泛的应用。
结果译文:
1.使用 ssDNA 文库制备进行的 cfDNA 分析揭示了先前未被认识的片段组学标志物
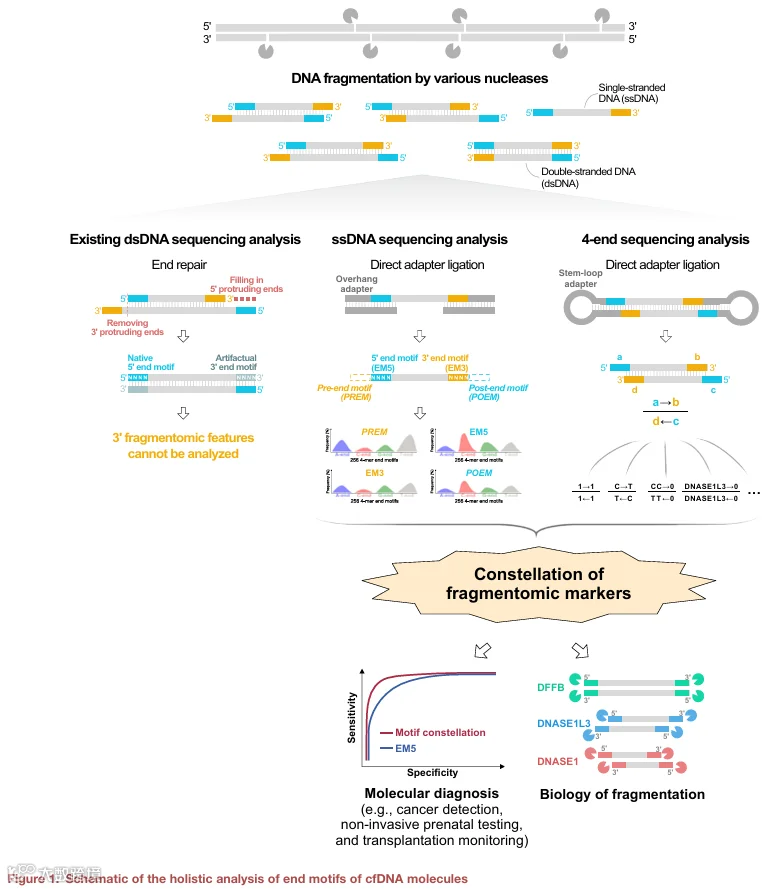
为了评估血浆 DNA 的片段大小和天然末端基序,我们采用了 ssDNA 文库制备,随后在 Illumina 平台上进行双端测序,在本研究中称为“2‑端测序”。简而言之,血浆 DNA 经过变性,将双链 DNA 分子转化为单链形式。得到的单链 cfDNA 片段直接连接测序接头,使 DNA 链能够单独测序(详见 STAR Methods)。这种 2‑端测序因此绕过了双链 DNA 文库制备所需的常规末端修复过程,保留了单个单链片段的天然末端。
我们分析了 38 名健康对照,中位数为 4600 万条双端读段(范围:2600 万至 5800 万条)。由 ssDNA 文库制备得到的血浆 DNA 大小分布显示出约 52 nt 和 166 nt 的两个明显峰,在这些峰之间可观察到微弱振荡的 10 nt 周期性。这一发现与先前几项报道一致。相比之下,对于双链 DNA 文库制备,仅在 166 bp 处出现一个主峰,对于低于 150 bp 的分子存在一系列 10 bp 的振荡(图 S1A)。在本研究中,当提及 ssDNA 时,使用单位“nt”。相反,当讨论 dsDNA 时,使用“bp”。
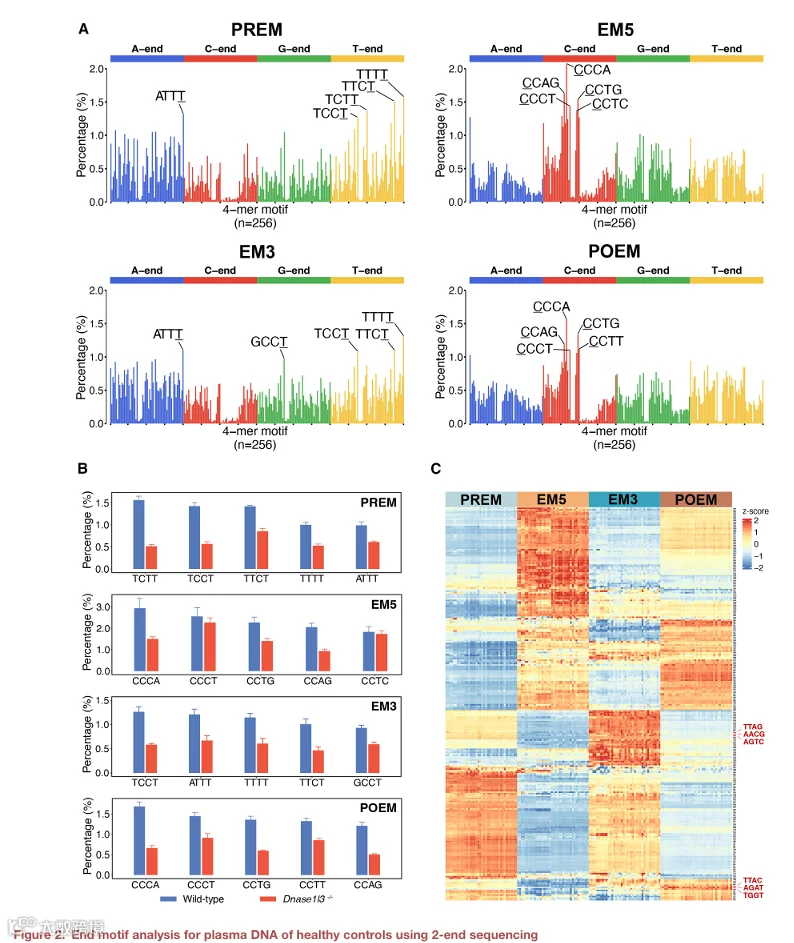
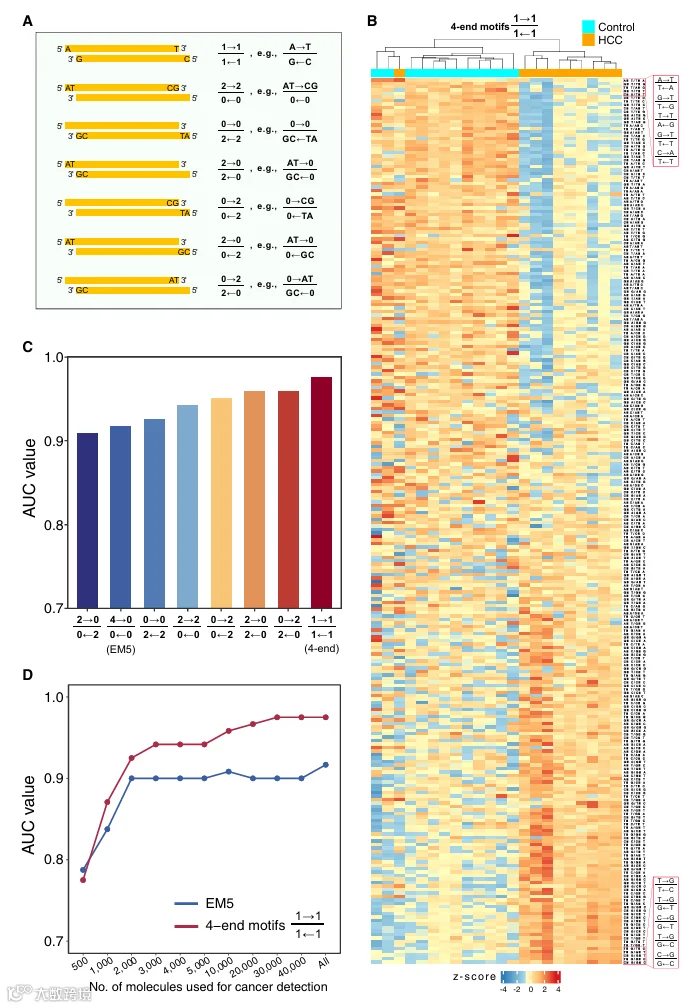
我们进一步研究了 cfDNA 的末端基序。我们假设负责切割基因组以产生片段化 cfDNA 分子的 DNA 核酸酶可能与切割位点侧翼的几个核苷酸相互作用。与测序末端紧邻的序列基序很可能参与了核酸酶与被切割 DNA 之间的相互作用。因此,我们研究了 EM5 和 EM3,以及从参考基因组推断的与 EM5 和 EM3 相邻的序列基序。位于测量的 EM5 之前的序列基序称为 PREM,位于 EM3 之后的基序称为 POEM(图 1)。在此分析中,我们按 5′ 到 3′ 方向确定了 PREM、EM5、EM3 和 POEM 的 4‑mer 末端基序,并计算了各自频率。
图 2A 显示了从 38 份健康人对照样本的测序结果中汇总的 PREM、EM5、EM3 和 POEM 分别的 256 种末端基序频率。256 种末端基序按字母顺序排列。以腺嘌呤(A)、胞嘧啶(C)、鸟嘌呤(G)和胸腺嘧啶(T)开头的基序分别以蓝色、红色、绿色和黄色突出显示。我们确定了 PREM、EM5、EM3 和 POEM 的前五种基序分别为(TTTT、TTCT、TCTT、ATTT、TCCT)、(CCCA、CCAG、CCTG、CCCT、CCTC)、(ATTT、ATTT、TTTT、TTCT、GCCT)和(CCCA、CCTG、CCAG、CCTT、CCCT)。4‑mer 基序以 5′ 到 3′ 方向书写,最靠近切割位点的碱基加下划线。例如,EM3 基序“ATTT”对应序列 5′‑ATTT‑3′,其中 3′ 端的末端核苷酸是“T”。值得注意的是,前几位的 EM5 和 POEM 主要以 5′ 端的胞嘧啶(C)开头。相比之下,前几位的 EM3 和 PREM 以 3′ 端的胸腺嘧啶(T)为特征,表明不同基序类别之间存在不同的切割偏好。PREM、EM5、EM3 和 POEM 排名前五的基序分别占总测序分子的 7.06%、8.03%、5.55% 和 6.29%。值得注意的是,这些前几位基序在 Dnase1/3⁻/⁻ 小鼠中与野生型小鼠相比均显著下调。另一方面,在 Dnase1⁻/⁻ 或 Dffb⁻/⁻ 小鼠中未观察到统计学显著变化(图 2B、S1B 和 S1C)。结果表明,这四类末端基序(PREM、EM5、EM3 和 POEM)主要由血浆中的 DNASE1L3 产生。
我们使用热图可视化了基于各自 4‑mer 末端基序的 PREM、EM5、EM3 和 POEM 的模式(图 2C)。PREM 和 EM3 具有明显的相似性,如热图中呈现的相似颜色模式所示。也观察到少数不一致的模式。对于 EM5 和 POEM 可以得出类似的结论,相似性超过了相对较小的差异。PREM 与 EM3 以及 EM5 与 POEM 的 Pearson 相关系数分别为 0.91 和 0.94(两者 p < 0.001),高于其他比较的 Pearson r 值(范围为 0.28 至 0.53)(PREM 与 EM5,0.28;PREM 与 POEM,0.46;EM5 与 EM3,0.36;以及 EM3 与 POEM,0.53)。我们发现,排名前 20 的基序在 PREM 和 EM3 之间以及 EM5 和 POEM 之间存在相当大的重叠,但在其他组合中则不然。在这些排名前 20 的基序中,EM5 和 POEM 之间共享 14 个,PREM 和 EM3 之间共享 18 个(表 S1)。除了 PREM 和 EM3 之间以及 EM5 和 POEM 之间大致相似的模式外,值得强调不一致的区域。例如,4‑mer 末端基序 TTAG、AACG 和 AGTC 在 PREM 和 EM3 之间的代表性存在差异,而 TTAC、AGAT 和 TGGT 在 EM5 和 POEM 之间显著不同(图 2C)。
我们进一步研究了末端基序模式如何与分别约在 52 nt 和 166 nt 出现的两个峰相关联(图 S1A)。我们选择了两个窗口,分别为 32–72 nt 和 146–186 nt,涵盖每个峰上下游各 20 nt。分析与这两个大小范围相关的分子的基序模式。当我们将热图应用于分析与大小分布中第一个峰相关的血浆 DNA 群体时,这四类末端基序的模式变得不太明显(图 S1D)。PREM 和 EM3 之间以及 EM5 和 POEM 之间的相关性分别降至 0.73 和 0.81。然而,对于与第二个峰相关的血浆 DNA 群体,与这四类末端基序相关的模式再次变得明显。EM5 与 POEM 之间以及 PREM 与 EM3 之间的 Pearson r 分别为 0.93 和 0.92(图 S1E)。此外,与大小在 70–166 nt(Pearson r,0.94 和 0.92)(图 S2B)和 166–600 nt(Pearson r,0.93 和 0.94)(图 S2C)的长 cfDNA 群体相比,EM5 与 POEM 之间以及 PREM 与 EM3 之间的 Pearson r 值在大小相对较短的 cfDNA 群体(42–70 nt)中较低(Pearson r,0.79 和 0.71)(图 S2A)。这些观察结果表明,长 cfDNA 群体在 EM5 与 POEM 之间以及 PREM 与 EM3 之间表现出更强的相关性,与第一个峰和第二个峰相关的大小范围中观察到的模式一致。
使用 EM5 末端基序的反卷积分析,我们发现 DNASE1L3 特征(例如 F‑profile I)在 cfDNA 浓度较高的个体中(中位数,38.9;范围,36.8–44.7)与浓度较低的个体相比(中位数,41.3;范围,35.9–44.8;p = 0.022,单侧 Mann‑Whitney U 检验)显著降低(图 S3A)。我们还观察到高浓度组中长 cfDNA 片段(>166 bp)比例较高的趋势(图 S3B),尽管未达到统计学显著性。这些发现总体上支持了 Malki 等人报道的 DNASE1L3 信号与 cfDNA 浓度之间的关系。
2.HCC 患者与非 HCC 患者血浆 cfDNA 的差异末端基序
为了探索本研究中鉴定的末端基序的诊断价值,我们使用 ssDNA 文库制备对 38 名健康对照、35 名慢性乙型肝炎病毒感染者但无 HCC 的患者以及 43 名 HCC 患者的样本进行了测序。我们获得了中位数为 4600 万条双端读段(范围,2100 万至 5800 万条)。
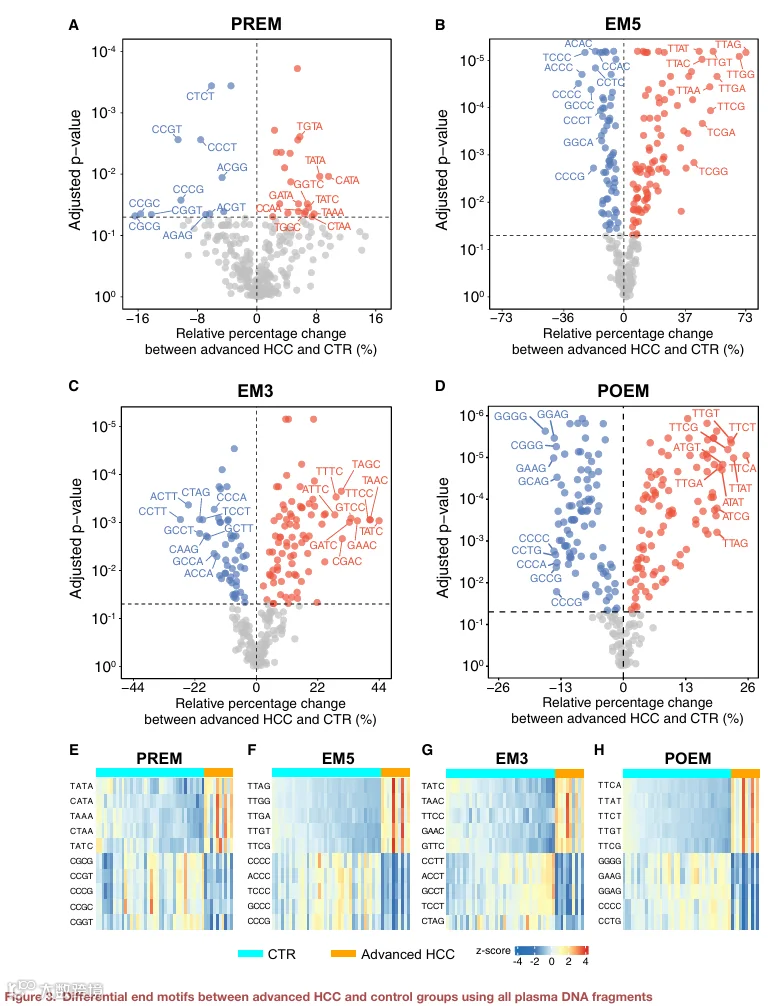
我们确定了晚期 HCC(即巴塞罗那临床肝癌分期 C 期)与健康对照组之间的差异末端基序(详见 STAR Methods)。如图 3A–3D 所示,当使用所有测序片段(即无计算机模拟大小选择)时,我们分别获得了与 PREM、EM5、EM3 和 POEM 相关的 34、158、139 和 194 个差异 4‑mer 末端基序。前五位差异基序在晚期 HCC 患者和对照之间的基序频率显示出明显模式(图 3E–3H)。对于 PREM,TATA、CATA、TAAA、CTAA 和 TATC 末端基序在晚期 HCC 组中表现出增加的代表性,而 CGCG、CCGT、CCCG、CCGC 和 CGGT 表现出减少的代表性(表 S2)。我们进一步确定了一个末端基序比率(EMR)指标,该指标通过计算在 HCC 组中表现出增加代表性的末端基序的累积频率与表现出减少代表性的末端基序的累积频率的比率得出。为所有样本(包括健康对照、HBV 携带者和不同肿瘤分期的 HCC 患者)确定了 EMR 值。HCC 组的 PREM EMR 显著更高(中位数,2.80;范围,2.54–3.56),与非 HCC 组相比(中位数,2.70;范围,2.38–2.96)(p < 0.0001;双侧 Mann‑Whitney U 检验)(图 S4A)。类似地,我们确定了 EM5(例如,增加的基序:TTAG、TTGG、TTGA、TTGT 和 TTCG;减少的基序:CCCC、ACCC、TCCC、GCCC 和 CCCG)、EM3(例如,增加的基序:TATC、TAAC、TTCC、GAAC 和 GTTC;减少的基序:CCTT、ACCT、GCCT、TCCT 和 CTAG)和 POEM(例如,增加的基序:TTCA、TTAT、TTCT、TTGT 和 TTCG;减少的基序:GGGG、GAAG、GGAG、CCCC 和 CCTG)的差异末端基序。4‑mer 基序以 5′ 到 3′ 方向书写,最靠近切割位点的碱基加下划线。对于这些类型的末端基序,我们还观察到与非 HCC 组相比,HCC 组的 EMR 值显著升高(图 S4B–S4D)。这些结果表明,这些鉴定的末端基序具有癌症检测的潜力。
3.多种末端基序的联合分析用于区分 HCC 患者与非 HCC 患者
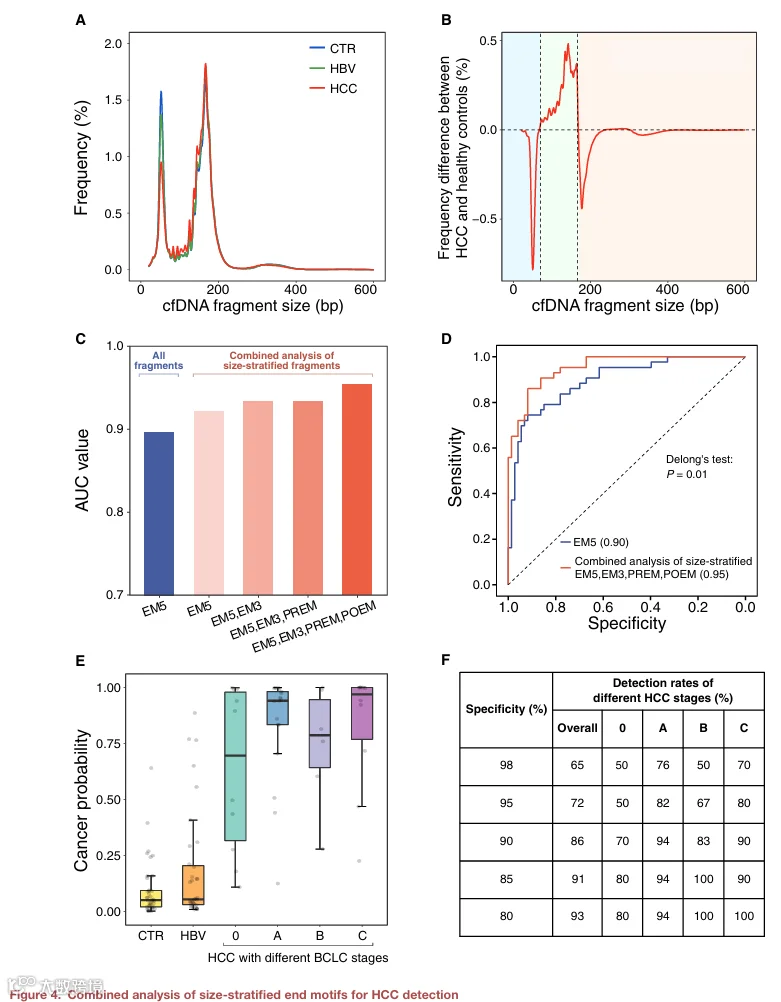
与 HBV 组和对照组相比,HCC 组中大小围绕第一个峰的片段相对量有所减少(图 4A)。具有最高肿瘤 DNA 分数(40%)的癌症患者与健康对照样本的中位大小谱之间的大小频率差异大致可分为三组(图 4B)。A 组(图 4B 中浅蓝色)在 42–70 nt 范围内表现出大小频率降低。B 组(图 4B 中浅绿色)在 70–166 nt 大小范围内显示出信号增加,随后对于大于 166 nt 的大小信号减少(C 组,图 4B 中浅红色)。我们比较了代表性癌症样本和健康对照样本之间的大小分布,以识别可能被视为癌症相关模式的特征性大小偏移。因此,在分析末端基序模式时利用有关大小范围的信息将是有用的。在 HCC 组和对照组的合并样本中也观察到总体上一致的大小频率差异(图 S4E)。
为了进一步探索本工作中鉴定的片段组学标志物的诊断潜力,我们开发了一个支持向量机模型,整合了所有这些标志物。该方法旨在利用来自 PREM、EM5、EM3 和 POEM 的 256 种 4‑mer 末端基序在三个不同大小范围内的独特特征,反映 HCC 和非 HCC 组之间潜在大小变化的影响。每个血浆 DNA 群体组为 PREM、EM5、EM3 和 POEM 分别贡献了 256 种 4‑mer 末端基序。因此,支持向量机进行癌症检测总共可以利用 3,072 个特征(4 种基序 × 256 种末端基序 × 3 个大小范围)。我们采用留一法评估诊断性能(详见 STAR Methods)。受试者工作特征曲线分析显示,来自所有片段的 EM5 得出的 AUC 为 0.90。此外,随着我们逐渐在三个大小范围内纳入更多片段组学标志物,AUC 持续提高,范围从 0.93 到 0.95(图 4C)。与传统的 EM5 分析相比,大小分层的末端基序联合分析使得癌症检测能力显著增强(p = 0.01,DeLong 检验)(图 4D)。使用联合特征,HCC 患者患癌概率显著高于对照组和 HBV 携带者(中位数,0.938 对比 0.053;范围,0.108–1.00 对比 0.000964–0.886;p < 0.0001,双侧 Mann‑Whitney U 检验)(图 4E)。我们通过改变特异性阈值检查了 HCC 检测的灵敏度,发现在特异性分别为 90%、85% 和 80% 时,HCC 的检出率分别为 86%、91% 和 93%(图 4F)。这些发现突出了全面整合本研究中鉴定的片段组学标志物的诊断潜力。此外,我们分别研究了使用 1‑、2‑、3‑、4‑ 和 5‑mer 末端基序的性能。结果显示,与其他 k‑mer 特征(AUC 范围,0.860–0.951)相比,4‑mer 末端基序(AUC,0.954)表现最佳(图 S5A)。
为了进一步验证模型的稳健性,我们进行了 1,000 次重采样的 Bootstrap 分析,以评估 AUC 的 95% 置信区间。我们将包括 EM5、EM3、PREM 和 POEM 的联合特征集与传统的基于 EM5 的 2‑端测序方法进行了比较。如图 S5B 所示,联合特征集在区分 HCC 患者与非 HCC 患者方面达到了 0.95(95% CI,0.94–0.97)的 AUC,显著高于仅 EM5 模型(AUC,0.90;95% CI,0.86–0.93;p < 0.01,基于 Bootstrap 的检验)。在 95% 的特异性下,联合模型的灵敏度为 0.70(95% CI,0.61–0.79),而仅 EM5 模型为 0.58(95% CI,0.45–0.76)。图 S5C 显示了精确率‑召回率曲线下面积,其中联合特征集再次优于仅 EM5 模型(AUPR,0.93 对比 0.84;95% CI,0.91–0.95 对比 0.80–0.90;p < 0.01,基于 Bootstrap 的检验)。这些 Bootstrap 结果支持模型性能的稳健性,减轻了关于潜在过拟合的担忧。
我们注意到非 HCC 组(中位数,63 岁;范围,26–72)与 HCC 组(中位数,68 岁;范围,44–82)相比倾向于稍年轻(p < 0.001,双侧 Mann‑Whitney U 检验)。我们在年龄匹配的组间重复了分类分析(图 S6A 和 S6B;详见 STAR Methods)。年龄匹配队列中 HCC 检测的性能与最初提交稿件中报道的性能相当(AUC,0.942 对比 0.954;p = 0.733,基于 Bootstrap 的检验)(图 S6C),表明年龄不太可能是我们研究中的一个重要混杂因素。
我们团队先前证明了利用 cfDNA 片段化模式推断 DNA 甲基化并进行癌症检测的可行性,称为基于片段组学的甲基化分析。然而,先前发表的 FRAGMA 技术主要关注 cfDNA 片段的 5′ 端(即 5′ FRAGMA)。我们想知道片段化模式与 DNA 甲基化之间的相关性是否也存在于 cfDNA 的 3′ 端(3′ FRAGMA)。
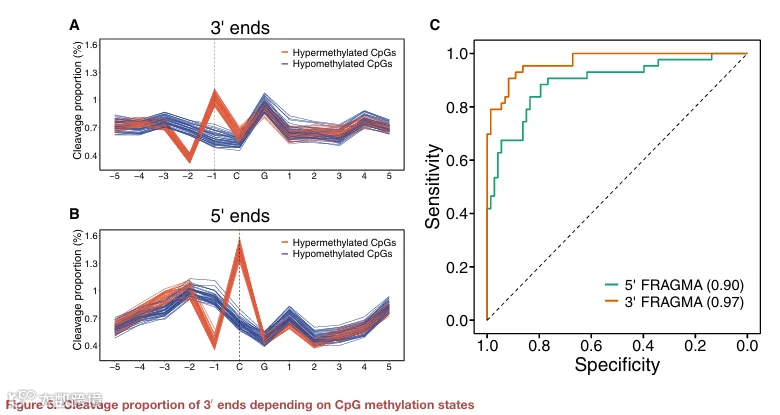
为此,我们将先前用于 5′ FRAGMA 的类似分析策略应用于分析 3′ FRAGMA。我们确定了如前所述的切割测量窗口内每个位置 3′ 端的切割比例。如图 5A 所示,在健康对照中,与高甲基化 CpG 位点相关的 3′ 切割模式与低甲基化 CpG 位点相关的模式显著不同。例如,与低甲基化 CpG 位点相关的群体相比(中位数,0.70 和 0.57),在与高甲基化 CpG 位点相关的 cfDNA 分子群体中,位置 -4 和 -1 表现出更高的切割比例值(中位数,0.75 和 1.03)。3′ 端最常终止于甲基化 CpG 位点前紧邻的 1 nt 位置。相比之下,对于 5′ 端,与高甲基化 CpG 位点相关的 cfDNA 分子群体中,CpG 位点的胞嘧啶处表现出比与低甲基化 CpG 位点相关的群体更高的切割比例值(中位数切割比例,1.45 对比 0.64)(图 5B)。5′ 端偏好于恰好位于甲基化 CpG 位点的胞嘧啶处,这与先前的发现一致。
为了评估使用 3′ FRAGMA 的诊断性能,我们采用支持向量机分析与差异甲基化 CpG 位点相关的 3′ 切割末端,以区分 HCC 患者与非 HCC 患者(详见 STAR Methods)。与对照组和 HBV 携带者相比,HCC 患者患癌概率显著更高(中位数,0.98 对比 0.02;四分位距,0.80–1.00 对比 0.003–0.17;p < 0.001,双侧 Mann‑Whitney U 检验)(图 S7)。这一发现表明,3′ FRAGMA 可用于测量 DNA 甲基化和检测癌症。值得注意的是,3′ FRAGMA 表现出优于 5′ FRAGMA 的性能,AUC 从 0.90 提高到 0.97(p < 0.01,DeLong 检验)(图 5C)。使用 0.32 的患癌概率阈值,特异性和灵敏度分别为 0.91 和 0.90。在 85% 和 80% 的特异性阈值下,相应的灵敏度均为 95%。
我们已经证明纳入 cfDNA 的 3′ 片段组学标志物有利于癌症检测。接下来,我们探索是否可能全息测定双链 cfDNA 分子的所有四个末端,并利用所有四个末端的片段组学信息进行癌症诊断。然而,2‑端测序的性质使得从测序结果中追踪双链分子的所有末端具有挑战性,因为原始的双链 cfDNA 分子在测序前已被分离成单链。为此,我们开发了一种新的测序方法来解决这个问题。
如图 S8 所示,双链 cfDNA 分子表现出各种末端形式,例如 3′ 突出端、5′ 突出端或平末端。这些 cfDNA 分子与茎环接头连接,这些接头具有定制的末端构型,能够与感兴趣的天然 cfDNA 分子杂交。例如,一个具有 5′ 突出链的 cfDNA 片段将连接一个匹配的茎环接头,该接头具有与突出端互补的精确长度的单链序列。末端信息,包括长度和碱基组成,被编码在接头茎区的序列条形码中(详见 STAR Methods)。因此,正确连接的分子形成闭合环状 DNA。采用核酸外切酶处理的额外步骤来消除不完全连接的产物,最大限度地减少错误连接导致条形码信息误读的风险。随后使用单分子实时测序对环化的 DNA 分子进行测序。在此方法中,同一分子的两条链被多次测序,从而能够解码单个双链分子的完整 4‑端信息(详见 STAR Methods)。我们将此方法称为 4‑端测序。
对于 4‑端测序,我们分析了 6 名对照、6 名 HBV 携带者和 10 名 HCC 患者的样本,中位数为 75,706 个分子(四分位距,46,377–166,786)。我们观察到 PREM、EM5、EM3 和 POEM 的中位频率总体上与 2‑端测序分析的结果相关(图 S9A–S9D),并且对于每一类末端基序,前 20 种基序在很大程度上重叠,两种技术之间平均有 65% 的重叠(表 S3)。然而,与其他类型的末端基序相比,EM3 的相关性似乎较低。2‑端和 4‑端测序之间观察到的差异可能源于技术的固有差异。例如,2‑端测序测量 ssDNA 和 dsDNA 分子,而 4‑端测序只能检测 dsDNA 分子。根据支持向量机分析,每种类型的末端基序在区分 HCC 和非 HCC 组方面都显示出一定程度的能力,AUC 值范围从 0.90 到 0.96(图 S9E)。
SMRT‑seq 可用的测序片段数量有限,这代表了首次实现 4‑端测序概念所面临的挑战。因此,作为概念验证研究,我们没有研究 4 个末端中每个末端的 4‑mer 末端基序,而是仅研究了每个末端的单核苷酸末端基序。我们还为此类 4‑端片段组学分析开发了一种特定的表示法。对于分析 4 个末端中每个末端的 1‑mer 基序,我们将此表示法记为 1−11−11−11−1。在此表示法中,Watson 链以 5′ 到 3′ 方向的两个末端放在分子中,方向由箭头指示。Crick 链的两个末端放在分母中。数字表示从末端开始的基序长度(例如,值为 1 对应 1‑mer 基序)。使用此表示法,分析一条链的 5′ 和 3′ 末端中每个末端的 2‑mer 基序将表示为 2−22−22−22−2 或 2−22−22−22−2。图 6A 显示了此表示法的使用示例。在某些应用中,显示特定基序的实际序列而不仅仅是其长度可能是有益的。因此,对于这些应用,可以明确说明实际的核苷酸序列(见图 6A 右侧)。例如,假设 Watson 链的 EM3 中存在末端 5′‑AT‑3′ 二核苷酸,Crick 链的 EM3 中存在末端 5′‑CG‑3′ 二核苷酸,则 4‑端基序可以表示为 ATCGCGAT。
对 256 种 4‑端基序频率的层次聚类分析显示,HCC 受试者明显聚集在一起,而非 HCC 受试者形成其他不同的簇(图 6B)。与对照组相比,HCC 组中减少最多的前 5 个 4‑端基序是 AAAA、AAAA、AAAA、AAAA 和 AAAA。相反,HCC 组中增加最多的前 5 个 4‑端基序是 CCCC、CCCC、CCCC、CCCC 和 CCCC。此外,我们利用支持向量机分析了 4‑端测序结果中定义的不同类型末端基序的分类能力。使用 4‑端基序在区分 HCC 患者与非 HCC 患者方面取得了最佳性能,AUC 达到 0.98,而其他类型的末端基序的 AUC 范围在 0.91 到 0.96 之间(图 6C)。
为了进一步研究测序片段数量如何影响 4‑端测序在癌症检测中的性能,我们进行了下采样分析,通过随机选择 500、1,000、2,000、3,000、4,000、5,000、10,000、20,000、30,000 和 40,000 个片段进行分类分析。图 6D 显示,随着测序片段数量的增加,4‑端基序分析的性能逐渐提高。例如,使用 1,000 个测序片段时,AUC 为 0.87,而使用 10,000 个测序片段时,AUC 增加到 0.96。当测序片段达到 20,000 时,性能达到平台期。值得注意的是,在分析的不同测序片段数量下,使用 4‑端基序分析在区分 HCC 患者与非 HCC 患者方面均显示出优于 EM5 分析的性能。即使使用比达到 0.95 AUC 所需的 4‑端基序分析多 4 倍的测序片段,EM5 分析仍未能达到相同的性能水平。数据表明,对双链 cfDNA 分子所有 4 个末端的全息分析提供了协同增强的诊断性能。
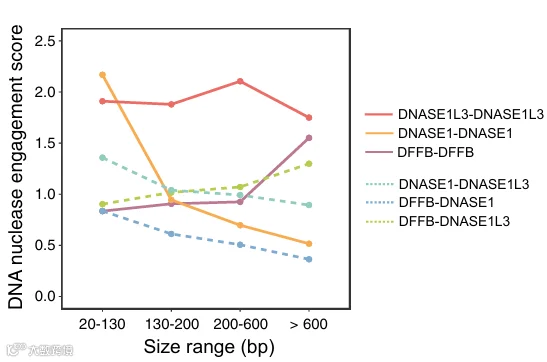
6.使用 4‑端测序对 cfDNA 核酸酶协调切割的生物学见解
使用核酸酶敲除小鼠和体外全血孵育实验已经揭示了核酸酶在 cfDNA 片段化中的作用。然而,血浆中的多种 DNA 核酸酶是否在 cfDNA 片段化过程中以协调方式作用仍然未知。为了解决这个问题,我们使用 4‑端测序来研究不同 cfDNA 分子群体中(根据片段大小)DNA 核酸酶的参与情况。我们根据野生型和核酸酶敲除小鼠之间的差异基序频率,将核酸酶特异的 5′ 末端基序和 3′ 末端基序定义为核酸酶切割特征(见 STAR Methods)。我们分析了每个片段同侧(即 2−02−02−02−0)的 5′ 端和 3′ 端的 DNASE1L3、DNASE1 和 DFFB 切割末端特征,分别形成 DNASE1L3‑DNASE1L3、DNASE1‑DNASE1和 DFFB‑DFFB特征。主要范围在 130 到 200 bp 的 cfDNA 片段以 DNASE1L3‑DNASE1L3 特征为特征,支持 DNASE1L3 作为 cfDNA 片段化的主要贡献者的作用(图 7)。有趣的是,DFFB‑DFFB 特征的丰度随片段大小增加而增加,特别是对于大于 600 bp 的片段,表明 DFFB 在 cfDNA 片段化的早期阶段起作用。相比之下,DNASE1‑DNASE1 特征的丰度在较短的 cfDNA 片段(大小范围,20–130 bp)中更高,在较长的片段中减少,表明 DNASE1 可能参与下游的片段化事件。DNASE1L3‑DNASE1L3 特征在广泛的片段大小范围内保持持续显著,表明 DNASE1L3 可能参与片段化的早期和晚期阶段。我们还观察到了混合切割模式,其中 cfDNA 片段同侧的 5′ 和 3′ 端与不同的核酸酶相关,例如 DNASE1‑DNASE1L3、DFFB‑DNASE1 和 DFFB‑DNASE1L3。在短分子(<130 bp)中,发现同质切割模式(例如 DNASE1L3‑DNASE1L3 和 DNASE1‑DNASE1)比异质切割模式(例如 DNASE1‑DNASE1L3)更频繁。类似地,在长分子(>600 bp)中,同质切割模式(例如 DNASE1L3‑DNASE1L3 和 DFFB‑DFFB)比混合特征(例如 DFFB‑DNASE1L3)更常见。这些发现表明,一旦 DNA 核酸酶在一条链上启动切割,它也可能协调随后在互补链上的邻近切割。这些发现证明了 4‑端测序在解析多种 DNA 核酸酶在 cfDNA 片段化中的动态和协调作用方面的能力,为这一生物过程的复杂性提供了新的见解。值得注意的是,在单链 cfDNA 文库中观察到的第一个峰并未出现在分析双链分子的 4‑端测序的大小分布中(图 S9F)。由于该平台通量较低,我们扩大了大小范围以确保有足够的数据用于末端基序分析。此处使用的大小分层与单链 cfDNA 分析中采用的分层不同。
4‑端测序的另一个特点是能够评估由协调切割产生的 cfDNA 的 5′ 和 3′ 锯齿状末端。值得注意的是,在先前发表的方法中无法分析 3′ 锯齿状末端。在本研究中,我们发现 Dnase1/3⁻/⁻ 小鼠中具有 3′ 锯齿状末端的片段数量显著减少(中位数:23.28% 和 9.79%)。敲除 DFFB 导致平末端减少,而 DNASE1 的缺失倾向于减少 5′ 锯齿状末端(图 S10)。
更多结果和补充图表:doi: 10.1016/j.xgen.2026.101142
长按二维码关注我们,用最短的时间和最高的效率学习更多数据分析方法!
扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:
https://cnsknowall.com
CNSknowall:24年最新问世的遥遥领先的科研数据(0代码生信+统计学)分析平台,同时含有机制图模块+汉化版Pubmed融合Deepseek高效筛选目标文献+SCI文献例句/语料检索模块+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动批阅!