胶质母细胞瘤(GBM)为何对免疫治疗“无动于衷”?答案藏在缺氧驱动的代谢重编程中!本研究通过纯公共数据库挖掘(TCGA、GEO等)结合空间转录组、单细胞RNA测序、代谢组学及MIBI-TOF多组学分析,首次系统揭示了GBM中缺氧-HIF-1α轴如何诱导IDO1在血管内皮细胞和髓系细胞中异常表达,导致色氨酸耗竭和犬尿氨酸堆积,进而通过GCN2-mTOR及AhR通路诱发CD8+ T细胞终末耗竭、Treg扩增,并促进VEGFA依赖性血管新生。文中还绘制了缺氧-犬尿氨酸代谢生态位的空间图谱,提出基于Kyn/Trp比值的预后生物标志物和联合靶向IDO1/AhR的精准免疫代谢治疗策略。全文干货,适合肿瘤免疫、生信分析及神经肿瘤学研究者收藏!
今天给大家解读一篇3月发表在《Metabolites》上的题目为“Nexus of IDO1/Kynurenine Pathway to T-Cell Exhaustion: Hypoxia-Induced Tryptophan Metabolism in Glioblastoma.”的文章。本文是一篇关于胶质母细胞瘤免疫代谢的综述。文章系统性地阐明了缺氧如何作为关键驱动因素,重塑GBM微环境中肿瘤细胞、髓系细胞、内皮细胞和淋巴细胞的代谢,重点聚焦于色氨酸-犬尿氨酸代谢轴。文章批判性评估了该通路导致T细胞耗竭、支持免疫抑制细胞扩增以及促进血管生成的分子机制,并基于空间转录组学、代谢组学和药理学研究,讨论了靶向该代谢轴以恢复抗肿瘤免疫的治疗策略及挑战。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!包括数据集、绘图代码、图表复现、思路总结、参考文献……0代码!鼠标点点点即可轻松完成5-10分生信SCI全文复现!
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!


团队成员合影(位于上海陆家嘴中心,可随时预约参观)
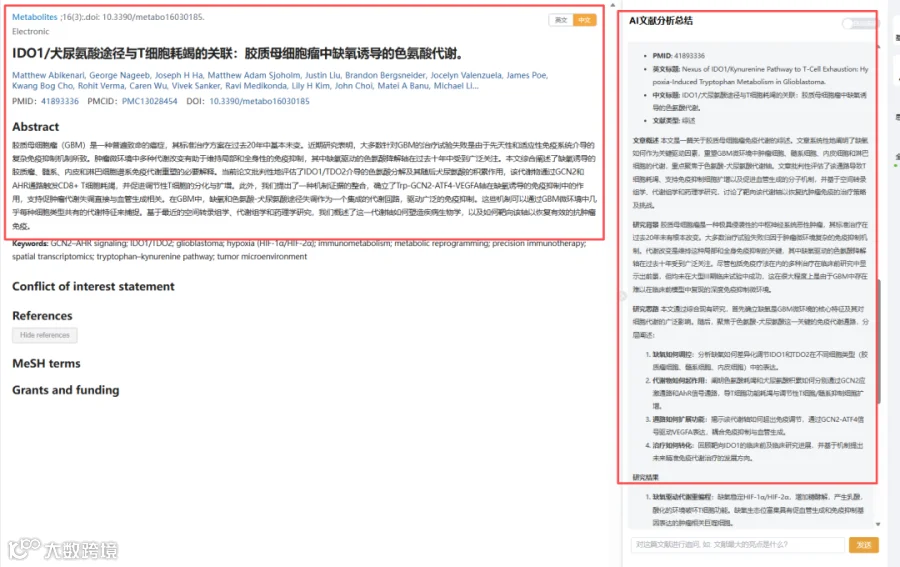
题目:《IDO1/犬尿氨酸途径与T细胞耗竭的关联:胶质母细胞瘤中缺氧诱导的色氨酸代谢》Nexus of IDO1/Kynurenine Pathway to T-Cell Exhaustion: Hypoxia-Induced Tryptophan Metabolism in Glioblastoma
发表期刊:Metabolites
影响因子:3.7
研究背景:
胶质母细胞瘤是一种极具侵袭性的中枢神经系统恶性肿瘤,其标准治疗在过去20年未有根本改变。大多数治疗试验失败归因于肿瘤微环境复杂的免疫抑制机制。代谢改变是维持这种局部和全身免疫抑制的关键,其中缺氧驱动的色氨酸降解轴在过去十年受到广泛关注。尽管包括免疫疗法在内的多种治疗在临床前研究中显示出前景,但均未在大型III期临床试验中成功,这在很大程度上是由于GBM中存在难以在临床前模型中复现的深度免疫抑制微环境。
CNSknowall 平台 Pubmed+AI 快速提炼全文要点
研究思路:
- 缺氧如何调控
分析缺氧如何差异化调节IDO1和TDO2在不同细胞类型(胶质瘤细胞、髓系细胞、内皮细胞)中的表达。
- 代谢物如何起作用
阐明色氨酸耗竭和犬尿氨酸积累如何分别通过GCN2应激通路和AhR信号通路,导T细胞功能耗竭与调节性T细胞/髓系抑制细胞扩增。
- 通路如何扩展功能
揭示该代谢轴如何超出免疫调节,通过GCN2-ATF4信号驱动VEGFA表达,耦合免疫抑制与血管生成。
- 治疗如何转化
回顾靶向IDO1的临床前及临床研究进展,并基于机制提出未来精准免疫代谢治疗的发展方向。
研究亮点:
- 空间异质性视角
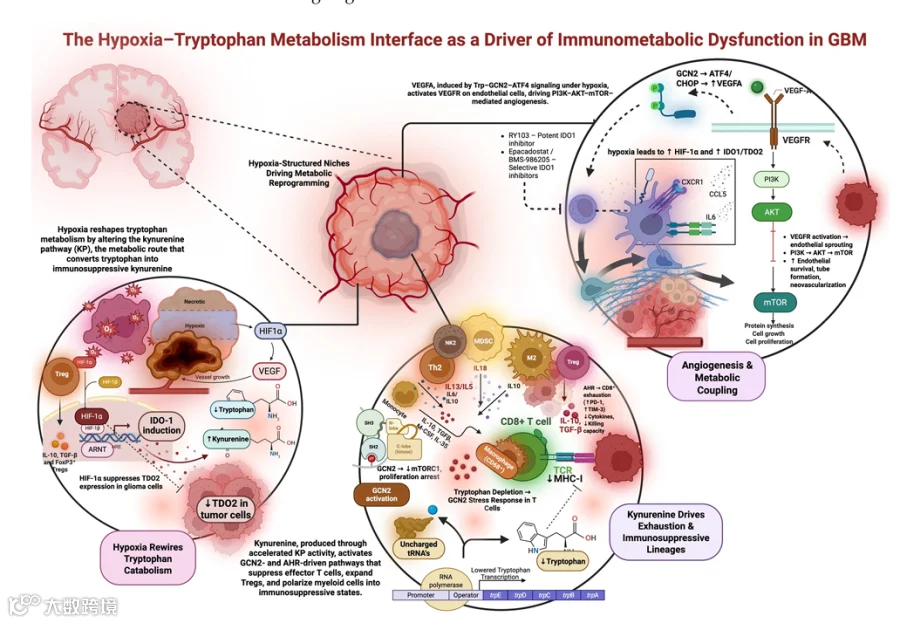
强调了缺氧与犬尿氨酸通路激活并非弥漫性发生,而是在特定的代谢/免疫生态位中共定位,该处聚集着耗竭的CD8+ T细胞、Tregs和促血管生成的内皮细胞。
- 代谢轴的双重作用
系统阐述了IDO1/犬尿氨酸途径不仅抑制抗肿瘤免疫,还通过Trp-GCN2-ATF4轴直接上调VEGFA,将免疫代谢与血管重塑偶联起来。
- 细胞类型特异性调控
揭示了缺氧对色氨酸代谢的调控具有细胞类型特异性(如在胶质瘤细胞中抑制TDO2,在抗原呈递细胞和内皮细胞中诱导IDO1),从而在空间上重塑了免疫抑制微环境。
研究结果:
- 缺氧驱动代谢重编程
缺氧稳定HIF-1α/HIF-2α,增加糖酵解,产生乳酸,酸化的环境破坏T细胞功能。缺氧生态位富集具有促血管生成和免疫抑制基因表达的肿瘤相关巨噬细胞。
- 色氨酸代谢的细胞特异性调控
在缺氧条件下,胶质瘤细胞的TDO2表达被抑制,而抗原呈递细胞和内皮细胞的IDO1表达被诱导。特别是肿瘤相关内皮细胞高表达IDO1,在血-瘤界面形成色氨酸匮乏、犬尿氨酸丰富的局部微环境,阻碍T细胞浸润。
- 导致T细胞耗竭的双重机制
- 色氨酸耗竭
激活T细胞内的GCN2激酶,抑制mTORC1通路,导致细胞周期阻滞和功能失能。
- 犬尿氨酸积累
作为AhR内源性配体,驱动CD4+ T细胞向Treg分化,并促使CD8+ T细胞上调PD-1等抑制性受体,进入耗竭状态。
- 驱动血管生成
IDO1介导的色氨酸分解激活GCN2-ATF4信号,直接转录上调VEGFA,促进血管新生。抑制IDO1可同时抑制免疫抑制功能和血管生成。
- 治疗策略与挑战
IDO1抑制剂(如依帕卡司他、吲哚莫德)在临床试验中显示出良好的安全性,但与放疗、替莫唑胺或PD-1抑制剂的联合疗效有待证实。疗效有限可能源于TDO2/AhR冗余、血脑屏障/血瘤屏障导致的递送问题以及肿瘤微环境的时空异质性。
研究总结:
本文得出结论:缺氧诱导的色氨酸代谢是GBM免疫抑制微环境的一个决定性轴心。它通过稳定HIF信号并将色氨酸分解代谢重新分配至内皮和髓系区室,影响了空间有序的生态位,从而选择性损害效应T细胞功能、维持抑制性髓系细胞状态,并将VEGFA依赖性血管生成与免疫逃逸代谢耦合。这表明GBM的演进不仅由遗传改变定义,更由肿瘤微环境中支配细胞行为的代谢回路所决定。
未来的治疗策略应基于精准免疫代谢的理念,结合代谢与免疫导向的干预措施。这需要:
- 利用空间多组学技术
(如空间转录组、MIBI-TOF成像)描绘“犬尿氨酸工厂”生态位,识别IDO1/KP高活性、缺氧且免疫排斥的患者亚型。
- 开发理性组合疗法
例如,将IDO1/AhR抑制剂或缺氧调节剂与免疫检查点阻断、抗血管生成治疗或放疗进行序贯或联合应用。
- 克服递送与异质性挑战
考虑使用纳米技术等新型递送系统,并设计能够同时靶向肿瘤、内皮和髓系区室冗余通路(如TDO2、AhR)的方案。最终,通过整合空间表型分析和生物标志物驱动的患者分层,有望将缺氧-色氨酸代谢轴的机制理解转化为GBM患者的临床获益。
结果译文:
缺氧是GBM TME的一个标志,由快速细胞更新、紊乱的血管结构和瘤内出血及坏死共同引起[25-28]。这些过程在TME中形成明显的氧梯度,在GBM TME内部建立结构改变。假栅栏状坏死是胶质母细胞瘤的常见组织病理学特征;它发生在微血管塌陷继发的缺氧损伤所驱动的细胞迁移部位[27,29,30]。
缺氧诱导因子(HIF-1α和HIF-2α)的稳定触发了GBM细胞代谢的协同转变。值得注意的是,最近的代谢测定已阐明,HIF-1α增加糖酵解速率,通过GLUT1和GLUT3促进葡萄糖转运,并抑制线粒体氧化磷酸化[31-34]。因此,ATP生物合成越来越依赖无氧代谢,产生乳酸。乳酸使细胞外环境酸化,进而破坏T细胞受体介导的信号传导,并抑制浸润性CD8+效应T细胞的细胞因子产生和增殖[31,35,36]。
也有证据表明,缺氧协调了髓系谱系的剧烈转变。单细胞转录组学和空间转录组学已证实,坏死周围的缺氧微生态位富含表达TREM2和SPP1的肿瘤相关巨噬细胞(TAMs),这些细胞抗原提呈活性减弱、基质重塑增强,且其基因表达程序与吞噬受抑和代谢适应性下降相一致[37-40]。这些TAMs高表达促血管生成基因,从而形成一个有利于肿瘤生长的免疫抑制微环境[23,32]。重要的是,这些过程并非静态的;乳酸和缺氧应激持续募集单核细胞并诱导转录组和代谢紊乱,从而产生这些抑制性TAM[34,41,42]。
在坏死核心的细胞微环境中,缺氧诱导色氨酸代谢。虽然HIF-1α可以抑制胶质瘤细胞中TDO2的表达,但在缺氧条件下,抗原提呈细胞和内皮细胞中IDO1的表达被诱导,从而在基质细胞中引发色氨酸分解代谢。在血-肿瘤界面表达IDO1的内皮细胞可以产生一个色氨酸贫乏、犬尿氨酸丰富的微环境,从而影响跨内皮免疫细胞的渗出,特别是先天髓系细胞(如中性粒细胞)向GBM微环境中的募集[23,43-45]。这种代谢转变有多种后果:GCN2应激反应激活、T细胞增殖抑制,以及通过AHR介导的转录重编程使T细胞分化为耗竭性和调节性T细胞。此外,KP活性显著增强并围绕缺氧形成模式化分布[12,46-48]。表1总结了缺氧-犬尿氨酸轴及其在胶质母细胞瘤中的免疫代谢后果。
更多近期机制研究进一步阐明了缺氧诱导色氨酸代谢之间的联系。IDO1介导的色氨酸分解代谢激活GCN2信号,进而促进VEGFA转录,这种营养物质的丧失驱动了血管生成。具体来说,我们的理解通过证据得到扩展,表明IDO1驱动的色氨酸耗竭直接促进血管重塑。这种Trp-GCN2-VEGFA回路将免疫代谢与血管重塑联系起来[10,14,46,51,52]。IDO1抑制导致GCN2激活受抑、VEGFA转录本表达减少并减少内皮管形成,从而支持这一解释。此外,IDO1过表达促进VEGFA表达、微血管CD34+微血管扩张和肿瘤生长,而抑制则消除了这些反应。这些数据表明,对VEGFA靶向治疗的抵抗可能至少部分是由于IDO1介导的代谢重编程,而不仅仅是VEGFA冗余[14,53-55]。这些相同的通路正在新兴的GBM试验中被靶向,其中CAR-T、NK细胞和髓系定向产品被设计来抵抗这种代谢耗竭的细胞状态。早期阶段结果显示,代谢和耐缺氧的细胞疗法优于未经修饰的对应疗法,这与上述机制相呼应[56-58]。
总之,缺氧在GBM内诱导了一个高度有序的免疫代谢抑制状态。当缺氧重新编程癌症的代谢环境时,它会诱导营养限制性应激,导致效应T细胞耗竭并促进特定的免疫抑制性髓系亚群。此外,缺氧通过诱导内皮细胞和髓系细胞中的IDO1,同步激活色氨酸分解代谢,从而增强犬尿氨酸通路信号。在此框架下,色氨酸耗竭激活GCN2和ATF4,转录上调VEGFA并促进肿瘤微环境内的血管生成程序。理解缺氧、免疫系统和代谢重编程之间的相互作用可以为战略性治疗干预奠定基础[9,46,49,59]。与GBM进展和免疫逃逸相关的关键缺氧诱导转录和代谢程序总结于表2。
2.1. 经典色氨酸(Trp)降解通路(IDO1、IDO2和TDO2)
GBM中的色氨酸分解代谢以犬尿氨酸通路为主,由三种双加氧酶启动:吲哚胺2,3-双加氧酶1和2(IDO1、IDO2)以及色氨酸2,3-双加氧酶(TDO2)[53,65,66]。这些酶催化Trp降解为N-甲酰犬尿氨酸(随后迅速转化为犬尿氨酸(Kyn))的第一步限速反应[58]。在正常生理条件下,TDO2在肝脏中组成性活跃以维持Trp稳态,而IDO1/IDO2在大多数组织中基础表达水平较低,但可被炎症细胞因子(如IFN-γ)快速诱导[67]。在GBM中,该通路常被上调。胶质瘤过表达IDO1/2和TDO2,导致局部Trp耗竭和Kyn代谢物积累[58]。值得注意的是,超过90%的GBM表达IDO1。重要的是,IDO1/2或TDO2的高水平与显著较短的生存期相关。机制上,Kyn作为芳香烃受体(AhR)的内源性配体,AhR激活使T细胞和巨噬细胞向免疫抑制表型倾斜。Kyn-AhR信号传导与免疫逃逸和促肿瘤效应均有关联。TDO2驱动的Kyn通过AhR依赖性上调水通道AQP4促进胶质瘤细胞迁移。因此,通过将Trp转向犬尿氨酸通路,GBM利用IDO1/IDO2/TDO2产生Kyn——一种肿瘤生长和免疫逃逸的强效介质[58,67,68]。
缺氧是Trp代谢的主要调节因子。低氧张力矛盾地抑制GBM细胞中TDO2的表达。最近一项研究表明,在缺氧条件下培养GBM细胞或化学稳定HIF-1α会导致一种可逆的、HIF1α依赖性的TDO2 mRNA和蛋白下调[69]。此外,GBM中的色氨酸代谢涉及肿瘤细胞、多种免疫细胞类型以及周围基质细胞群(尤其是髓系免疫细胞和血管周围细胞)之间的串扰[70,71]。免疫细胞产生的炎症信号可以上调其他免疫区室中的IDO1/TDO2,而肿瘤代谢物则影响免疫和内皮细胞功能。例如,浸润T细胞释放的IFN-γ和其他细胞因子驱动肿瘤细胞和免疫细胞中的IDO1表达[60,72]。
缺氧并非以相同方向或相同程度均匀调节胶质母细胞瘤微环境中的色氨酸分解代谢。相反,缺氧具有细胞类型特异性效应,在功能上将Trp分解代谢活性在肿瘤和基质之间重新定向。对于胶质瘤细胞,HIF-1α积累可能抑制TDO2表达,这是一种恶性肿瘤细胞表型特征性的O2依赖性转录反应。然而,这并不意味着Trp-Kyn通路被全局抑制,因为相同的缺氧环境也富含驱动IFN-γ细胞因子网络激活的信号,而IFN-γ已知可调节抗原提呈细胞类型以及位于血-肿瘤界面的特化内皮细胞类型中的IDO1表达。因此,缺氧抑制TDO2活性和炎症细胞因子调节IDO1表达并非描述同一现象的相互矛盾的说法,而是两种不同的过程,在胶质瘤微环境中的不同细胞类型中同时发生[14,55,69,72]。
功能上,结果是免疫抑制性Trp分解代谢速率增加,无论胶质瘤细胞类型中TDO2活性如何降低。这种Trp分解代谢活性转移的后果是形成了高度局部化但极其强效的色氨酸耗竭、犬尿氨酸富集的微环境,由于它们发生在GBM基质和浸润性免疫细胞类型之间的界面上,因此在施加应激反应(GCN2-mTOR抑制)以及犬尿氨酸-AhR依赖性T细胞表型耐受重编程方面非常高效,无论缺氧环境中特征性的TDO2活性降低如何。因此,缺氧并没有将Trp分解代谢的位置(缺氧环境中TDO2活性较低)与总体结果(色氨酸耗竭、犬尿氨酸介导的应激和耐受重编程)分开,同时保持或强化了免疫抑制表型——这正是本综述作为胶质母细胞瘤免疫环境关键组成部分的核心主题[31,32,60,66]。
最后,血-肿瘤边界处内皮细胞表达IDO1不仅仅是一种相关性,而是可能充当免疫微环境中的一个主动过滤器,在淋巴细胞试图向肿瘤内外渗的部位创造一个色氨酸低、犬尿氨酸高的局部环境。
此外,对人GBM进行单细胞RNA测序发现,IDO1主要由肿瘤相关内皮细胞(ECs)表达,集中于一个共表达趋化因子CXCL11及下游JAK/STAT通路基因的细胞亚群。肿瘤细胞通常仅在典型的促炎细胞因子刺激下才表现出微弱的IDO1表达,而IDO1+内皮细胞簇则在血-肿瘤界面形成了一个局部的色氨酸耗竭、犬尿氨酸富集的微生态位[73]。GBM中这种内皮IDO1活性可通过阻碍T细胞浸润和功能,促成免疫豁免微环境[63]。
髓系来源的瘤内细胞,包括常驻小胶质细胞、单核细胞来源的巨噬细胞和髓系来源抑制细胞(MDSCs),既影响Trp代谢,也对其做出响应。这些细胞可以表达Trp分解代谢酶,并产生免疫抑制性代谢物(如乳酸和腺苷)作用于邻近细胞[67]。一个肿瘤-髓系代谢串扰的典型例子发生在IDH突变胶质瘤中。肿瘤细胞分泌促癌代谢物R-2-羟基戊二酸(R-2-HG),通过强力增强浸润性巨噬细胞的TDO2酶活性来重编程这些巨噬细胞。反过来,巨噬细胞从Trp大量产生犬尿氨酸,从而激活肿瘤微环境中的AhR。这种重编程通过使巨噬细胞向抑制性(M2样)状态倾斜,支持Treg并抑制CD8+ T细胞活性,从而诱导免疫抑制,强化了犬尿氨酸富集的生态位。
值得注意的是,TDO2缺陷的巨噬细胞对R-2-HG诱导的T细胞耗竭效应具有抵抗性[67]。总之,这些相互作用支持了GBM中合作代谢的影响:肿瘤细胞、免疫细胞和内皮细胞共同调节Trp-犬尿氨酸通路,以维持免疫耐受,并允许GBM特征性的持续局部和全身免疫抑制。通过共享酶工作量和代谢物交换,肿瘤细胞确保色氨酸耗竭和犬尿氨酸副产物持续存在——无论是在常氧区通过肿瘤自身的TDO2活性,在侵袭边缘通过IDO1+巨噬细胞和小胶质细胞,还是在肿瘤血管生态位通过IDO1+内皮细胞。这种代谢串扰最终支持肿瘤免疫逃逸,突出了GBM中多种细胞类型作为中断色氨酸代谢疗法的潜在靶点。表3描述了胶质母细胞瘤中色氨酸-犬尿氨酸通路的主要改变及其下游免疫效应。
3.1. T细胞耗竭的机制
GBM的特征在于其免疫抑制性肿瘤微环境,该微环境驱动效应T细胞功能障碍并最终耗竭[77,87,88]。GBM中的肿瘤浸润淋巴细胞(TIL)通常表达多种抑制性免疫检查点,如PD-1、TIM-3和LAG-3,这与耗竭T细胞表型一致。这些受体在CD8+ T细胞上的共表达与效应功能和增殖受损相关,尽管存在抗原[88]。与此同时,GBM施加代谢限制,使T细胞缺乏能量。其中一个例子是胶质瘤和髓系细胞上调IDO1,导致Trp快速耗竭。Trp对T细胞生长和功能至关重要[61]。此外,IDO1驱动的色氨酸耗竭激活T细胞中的应激反应激酶GCN2,抑制mTOR信号,导致效应T细胞细胞周期停滞和无能[62,64]。
Opitz等人首次将Kyn鉴定为胶质瘤中的内源性AhR配体,将色氨酸代谢与促进免疫耐受的基因表达程序联系起来[6]。Kyn进入T细胞结合AhR,触发转录程序,使T细胞向调节性和耗竭状态倾斜[78]。在CD4+ T细胞中,AhR激活诱导转录因子FoxP3,使这些细胞以效应表型为代价分化为Treg[12]。在CD8+ T细胞中,Kyn-AhR信号上调抑制性受体(如PD-1),同时抑制IL-2产生,从而强化耗竭[47,89]。这种分子重编程是导致T细胞耗竭、削弱GBM中抗肿瘤T细胞活性的关键机制。
3.3. 犬尿氨酸介导的Treg和MDSCs扩增的证据
GBM肿瘤微环境的一个标志是免疫抑制性细胞的丰富存在,特别是FoxP3+ Treg和MDSCs[68,90]。犬尿氨酸通路积极驱动这些细胞的扩增和募集。在临床样本中,高IDO1与Treg浸润增加和患者预后较差相关,表明色氨酸分解代谢与Treg介导的免疫抑制相关[61,68]。在临床前小鼠肿瘤模型中,IDO1过表达促进未成熟髓系细胞的扩增,这些细胞在浸润Treg的影响下分化为免疫抑制性MDSCs[50]。AhR信号诱导CCR2和其他趋化因子受体,促进髓系细胞向Kyn富集的肿瘤区域迁移[74]。总之,这些观察结果表明Kyn信号扩增了GBM中两个主要的免疫抑制性细胞群,最终塑造了其特征性的对有效抗肿瘤T细胞反应难治的微环境。图1概括了缺氧驱动的色氨酸代谢重编程如何导致胶质母细胞瘤中的免疫抑制和血管重塑。
破坏IDO1活性已被研究作为GBM的治疗策略。在可用的IDO1抑制剂中,epacadostat、indoximod、PF-06840003和BMS-986205受到了特别关注[91]。Indoximod作为色氨酸模拟物,破坏由IDO1活性驱动的下游免疫抑制信号[92]。值得注意的是,indoximod(D-1-甲基-色氨酸)不是直接的IDO1酶抑制剂;而是通过功能性模拟色氨酸,在IDO介导的色氨酸耗竭下游发挥作用,以恢复T细胞中的mTORC1信号,从而对抗饥饿诱导的免疫抑制。这种下游mTORC1救援机制在生物学上不同于直接的IDO1酶阻断(如epacadostat),这可能有助于解释为何在通路旁路策略与试图从酶源头完全抑制犬尿氨酸产生的尝试之间,临床反应可能不同。
在一项评估安全性和初步疗效的1期试验(NCT02502708)中,indoximod耐受性良好,在16名GBM患者中有1名出现部分缓解[93]。随后的1/2期试验(NCT02052648)进一步证明,indoximod仍然耐受性良好,包括与替莫唑胺(TMZ)和立体定向放疗联合给药时[94]。正在进行的试验继续扩大其临床评估:一项1期研究(NCT05106296)正在招募患者,以测试indoximod联合布鲁顿酪氨酸激酶(BTK)抑制剂加化疗;而一项2期试验(NCT04049669)正在入组儿童胶质瘤(包括GBM)患者,评估其与放化疗联合使用的情况[75,95]。
Epacadostat通过竞争性抑制直接降低IDO1酶活性[81,92]。在一项1/2期试验(NCT02327078)中,epacadostat联合PD-1阻断显示出良好的安全性[82,96]。一项2期临床试验(NCT03532295)目前正在评估epacadostat联合PD-1抑制、放疗和血管内皮生长因子(VEGF)阻断用于复发性胶质瘤。初步发现表明,这种多模式方案也是耐受性良好的[76]。
PF-06840003和BMS-986205是另外两种已进入早期临床评估的小分子IDO1抑制剂。PF-06840003的安全性在1期试验NCT02764151中进行了评估。尽管该研究最终由申办方终止,但初步数据显示出良好的安全性[97,98]。BMS-986205目前正在一项活跃的1期试验(NCT04047706)中与PD-1阻断联合进行研究,但在撰写本综述时尚未报告初步结果[79]。尽管越来越多的IDO1靶向药物在临床开发中取得进展,但不同研究中疗效的差异凸显了需要能够识别最可能从IDO1抑制中获益的患者的稳健生物标志物。
在临床前研究中,纳米技术为向GBM递送抗IDO疗法提供了一个有前景的平台。PPRX-1701是一种载有6-溴靛玉红-3'-乙酰氧肟的可生物降解聚合物,已被证明可以下调GBM细胞中的IDO1表达。在免疫活性小鼠GBM模型中,PPRX-1701治疗显著延长了生存期并重塑了肿瘤微环境,增加了CD8+ T细胞浸润,同时减少了免疫抑制性髓系细胞群[80]。基于纳米颗粒的递送也已应用于IDO1的基因靶向。例如,一种多功能包膜型纳米装置(MEND)已被用于将IDO1特异性siRNA递送到树突状细胞中,实现了有效的基因敲低和IDO1表达降低[99]。尽管基于纳米技术的GBM方法仍处于早期开发阶段,但成功的临床转化将取决于证明其安全性、可扩展性以及相对于传统药物递送方法的明确优势。尽管如此,令人鼓舞的临床前结果为持续创新抗IDO疗法的纳米尺度递送提供了强有力的依据。
为了更好地对患者进行分层和定制治疗,研究人员正在识别反映GBM中IDO1通路活性的生物标志物。犬尿氨酸/色氨酸(Kyn/Trp)比值是一种基于血液的代谢指数,由1-犬尿氨酸和1-色氨酸的水平计算得出,该测量值有望作为诊断或预后工具以及用于监测治疗反应。它是IDO/TDO酶活性的替代指标,较高的IDO1/TDO活性会将更多的Trp转化为Kyn。在癌症患者中,升高的Kyn/Trp比值通常表示免疫抑制性肿瘤微环境。具体在GBM中,研究表明与健康对照相比,患者的Kyn/Trp比值显著更高[70,85]。临床上,术后高Kyn/Trp比值与较短的总生存期相关,强调了其潜在的预后价值[100]。
总体而言,IDO1抑制在多项临床试验中显示出良好的安全性,即使与高细胞毒性治疗(如替莫唑胺、放疗或免疫检查点阻断)联合给药也是如此。尽管耐受性令人鼓舞,但临床反应凸显了需要阐明IDO1抑制是否能与其他治疗方式(包括免疫治疗、靶向治疗和基于纳米技术的递送系统)产生有意义的协同作用。在最近一项针对复发性胶质母细胞瘤患者的2期试验中,retifanlimab(抗PD-1)联合低分割放疗和贝伐珠单抗显示出有希望的总生存率(OS-9为71.4%),且无剂量限制性毒性,这支持了免疫治疗与细胞毒性和抗血管生成治疗联合使用的良好安全性。由于积极的安全性特征,随后加入了一个包含IDO1抑制剂epacadostat的队列,探索性免疫学研究仍在进行中。这些试验证实,IDO1抑制剂在联合方案中的安全性已得到确认,但尚未确定在哪些情况下IDO1抑制剂治疗能提供额外获益[100]。
IDO1抑制的有限疗效可能不仅仅源于患者选择因素:时机和顺序可能至关重要,因为Trp和Kyn轴可能变得嵌入慢性免疫功能障碍中。单药治疗也可能因TDO2和AhR冗余以及肿瘤微环境中关键通路抑制不足而被绕过。这些机制现实凸显了需要测试联合疗法,例如IDO1抑制剂与检查点阻断剂、血管调节剂以及专门设计用于解决空间异质性的放疗顺序联合。
持续的研究,特别是通过生物标志物驱动的患者选择和合理的联合策略,对于确定IDO1靶向药物能否将其强有力的临床前理论基础转化为GBM患者一致的临床获益至关重要。针对胶质母细胞瘤中缺氧-犬尿氨酸共激活的治疗策略的结构化概述见表4。
最后,靶向GBM中IDO1-犬尿氨酸轴的一个关键转化障碍在于,即使是生物学上合理设计的代谢疗法,当递送和空间背景未得到充分解决时也可能失败。与许多其他癌症不同,GBM肿瘤微环境通过血脑屏障(BBB)以及可变的血-肿瘤屏障与血流隔离,因此化合物的全身递送绝不能保证足够的肿瘤浓度,尤其是在肿瘤微环境的浸润性肿瘤区域,那里存在免疫-血管相互作用和IDO1+内皮细胞生态位。与此同时,GBM的肿瘤微环境高度异质,IDO1/TDO2酶的表达以及犬尿氨酸通路的活性在肿瘤、内皮和髓系细胞区室中差异分布,因此即使通路在肿瘤微环境的某些区域得到充分抑制,在其他区域仍可能存在免疫逃逸区域(如TDO2/AhR冗余通路)。
当前的一个核心挑战是摆脱“一刀切”的IDO1阻断策略,转向GBM的精准免疫代谢靶向治疗。近期基于批量测序和单细胞数据集的研究显示,IDO1、TDO2及下游KP酶在不同胶质瘤亚型中呈异质性表达,且KP活性与免疫抑制性髓系状态及T细胞功能障碍密切相关[40,57]。新兴临床数据同样表明,高表达IDO1/TDO2或AhR以及高Kyn/Trp比值与较短生存期相关,提示它们是可操作的生物标志物[65,83,91]。因此,未来研究应定义整合KP基因/蛋白表达、缺氧特征、全身性Kyn/Trp比值以及T细胞耗竭和髓系极化的功能性读数的免疫代谢“内分型”。具有KP高、AhR活跃、缺氧肿瘤的患者可以被前瞻性地分配到IDO1/TDO/TDO2抑制剂、AhR拮抗剂或效应T细胞代谢支持联合PD-1/PD-L1阻断的方案中,而KP低的肿瘤则可转向替代免疫疗法。
实现这样的患者筛选需要解析色氨酸代谢和缺氧在何处、何种细胞中协同抑制免疫。多重离子束成像平台(如MIBI-TOF)已经能够在实体瘤中以亚细胞分辨率绘制数十种蛋白的空间分布,并揭示出可预测预后的高度结构化的肿瘤-免疫微环境[84,101,102]。将MIBI-TOF或相关高多重成像技术应用于GBM,并采用靶向IDO1/TDO2、KP酶、缺氧标志物(HIF-1α、CA9)、色氨酸转运蛋白、免疫检查点配体以及谱系/功能标志物的抗体组合,将能够直接可视化“犬尿氨酸工厂”及其与耗竭CD8+ T细胞、Tregs、抑制性TAM/MDSC状态之间的关系。与此同时,单细胞转录组学和空间转录组学可以量化不同微生态位中的KP和缺氧基因程序,将其与T细胞受体(TCR)克隆型相关联,并追踪这些结构在放疗、抗血管生成治疗或IDO1/AhR靶向药物作用下的重塑过程[67,86,103,104]。这些整合的空间图谱将提供机制上可靠的生物标志物(例如KP高表达的缺氧微生态位),直接用于临床试验设计。
最后,缺氧必须被视为IDO1生物学中的共同驱动因素,而不仅仅是背景因素。临床前研究表明,缺氧稳定HIF-1α、上调PD-L1、重塑髓系细胞区室,并可直接诱导抗原提呈细胞中的IDO1表达,这提示放疗或抗血管生成方案可能加剧缺氧,从而增强KP介导的免疫抑制[103,105,106]。因此,试验方案应测试合理的序贯策略,例如先行IDO1/AhR阻断或缺氧调节药物,随后进行放化疗,并明确监测缺氧和KP活性如何共同演变。此外,在多种中枢神经系统肿瘤中,一个统一的主题正变得清晰:代谢功能(而非孤立的基因突变)主导着免疫抑制、血管重塑和增殖行为。这些相似性提示,靶向肿瘤细胞与免疫细胞之间共有的代谢应激通路,可能比单独使用通路特异性或受体特异性策略更为有效。将精准免疫代谢学原则(如空间图谱绘制和生物标志物锚定的试验设计)整合起来,为我们将缺氧诱导的色氨酸代谢日益深入的机制理解转化为GBM患者持久的临床获益提供了一条路径。最后,大语言模型和肿瘤学多模态AI的进展带来了一个现实的前景:将空间组学与数字病理学整合,在患者水平预测免疫治疗的反应和耐药模式,从而在中枢神经系统肿瘤中更精确地部署免疫代谢干预措施[107]。此外,激素受体通路,如雌激素和孕激素信号,已知会汇聚于PI3K/AKT/mTOR信号,后者是葡萄糖摄取、脂质合成和合成代谢的主要控制器。因此,用于治疗脑膜瘤的内分泌药物可以作为代谢通路调节剂在GBM中重新利用,以抑制上游生长因子/激素介导的mTOR信号[108-111]。尽管PIK3CA突变不是GBM的主要驱动因素,但PI3K信号在GBM中通过EGFR信号/PTEN缺失而被过度激活。将内分泌治疗与PI3K/mTOR通路药物联合使用,以靶向代谢适应性而非单一靶向细胞死亡的单药策略,是转化为GBM治疗的一个合理依据[81,112,113]。
更多结果和补充图表:doi: 10.3390/metabo16030185
长按二维码关注我们,用最短的时间和最高的效率学习更多数据分析方法!
扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:
https://cnsknowall.com
CNSknowall:24年最新问世的遥遥领先的科研数据(0代码生信+统计学)分析平台,同时含有机制图模块+汉化版Pubmed融合Deepseek高效筛选目标文献+SCI文献例句/语料检索模块+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动批阅!