今天给大家解读一篇3月发表在《The Journal of Clinical Investigation》上的题目为“SHMT2 deficiency disrupts transcriptional regulation through homocysteine-mediated suppression of histone lactylation in Huntington's disease models.”的文章。本研究通过整合RNA测序和代谢组学分析,发现HD中存在一碳代谢失调,并聚焦于SHMT2的下调。功能实验表明,SHMT2抑制加剧突变亨廷顿蛋白聚集和神经元退化,而过表达则改善病理表型。机制上,SHMT2缺乏导致HCY积累,进而抑制组蛋白乳酸化并扰乱转录调控。此外,HD药物氟哌啶醇被证明可调节这一通路,提示代谢-表观遗传轴作为治疗靶点。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!包括数据集、绘图代码、图表复现、思路总结、参考文献……0代码!鼠标点点点即可轻松完成5-10分生信SCI全文复现!
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!









团队成员合影(位于上海陆家嘴中心,可随时预约参观)





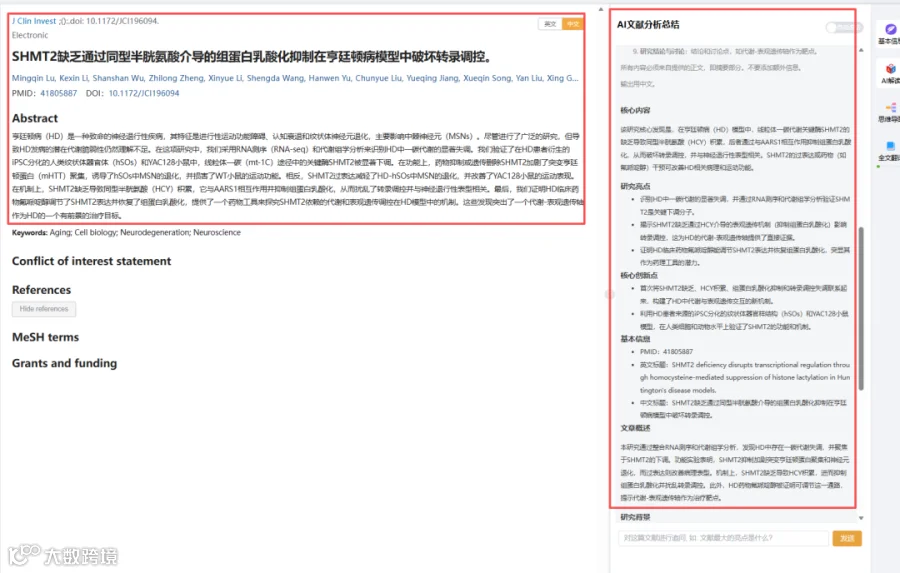
题目:《SHMT2缺乏通过同型半胱氨酸介导的组蛋白乳酸化抑制在亨廷顿病模型中破坏转录调控》SHMT2 deficiency disrupts transcriptional regulation through homocysteine-mediated suppression of histone lactylation in Huntington's disease models
发表期刊:The Journal of Clinical Investigation
影响因子:13.6
研究背景:
亨廷顿病(HD)是一种致命的神经退行性疾病,以进行性运动功能障碍、认知衰退和纹状体神经元(尤其是中棘神经元)退化为特征。尽管已有广泛研究,但导致HD发病的代谢脆弱性仍不清楚。
研究思路:
- 研究采用RNA测序和代谢组学分析识别HD中的代谢失调,并验证SHMT2在HD模型(人源iPSC分化的纹状体器官样结构和YAC128小鼠)中的表达变化。
- 通过药理学抑制、遗传删除或过表达SHMT2,评估其对突变亨廷顿蛋白聚集、神经元退化和运动功能的影响。
- 机制上,探讨HCY积累如何与AARS1相互作用抑制组蛋白乳酸化,从而影响转录调控,并使用氟哌啶醇作为工具验证SHMT2依赖的调节。
研究亮点:
-
识别HD中一碳代谢的显著失调,并通过RNA测序和代谢组学分析验证SHMT2是关键下调分子。 -
揭示SHMT2缺乏通过HCY介导的表观遗传机制(抑制组蛋白乳酸化)影响转录调控,这为HD的代谢-表观遗传轴提供了直接证据。 -
证明HD临床药物氟哌啶醇能调节SHMT2表达并恢复组蛋白乳酸化,突显其作为药理工具的潜力。
研究结果:
-
SHMT2在HD患者来源的纹状体器官样结构和YAC128小鼠中显著下调。 -
抑制或删除SHMT2加剧突变亨廷顿蛋白聚集,诱导中棘神经元退化,并损害野生型小鼠运动功能;相反,过表达SHMT2减轻神经元退化并改善YAC128小鼠运动表现。 -
SHMT2缺乏导致HCY积累,HCY与AARS1相互作用并抑制组蛋白乳酸化,进而扰乱转录调控,与神经退行性表型相关。 -
氟哌啶醇能调节SHMT2表达并恢复组蛋白乳酸化。
研究总结:
结果译文:
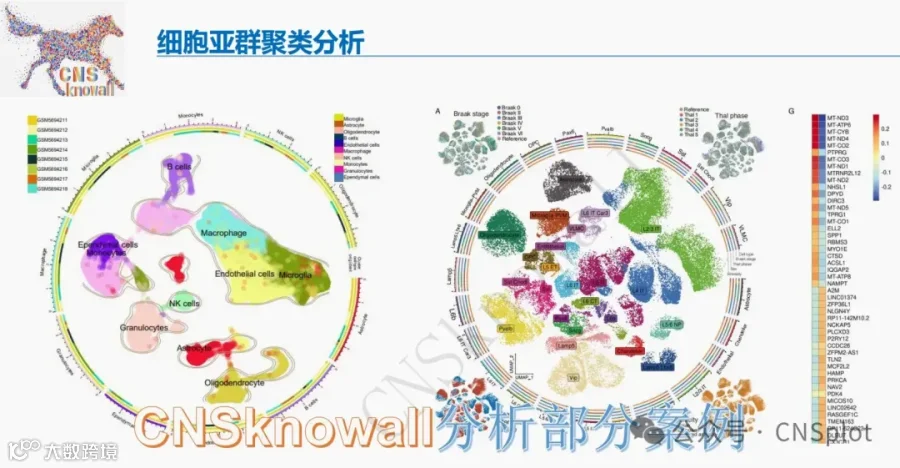
1.多组学分析揭示HD模型中一碳代谢失调和SHMT2表达降低
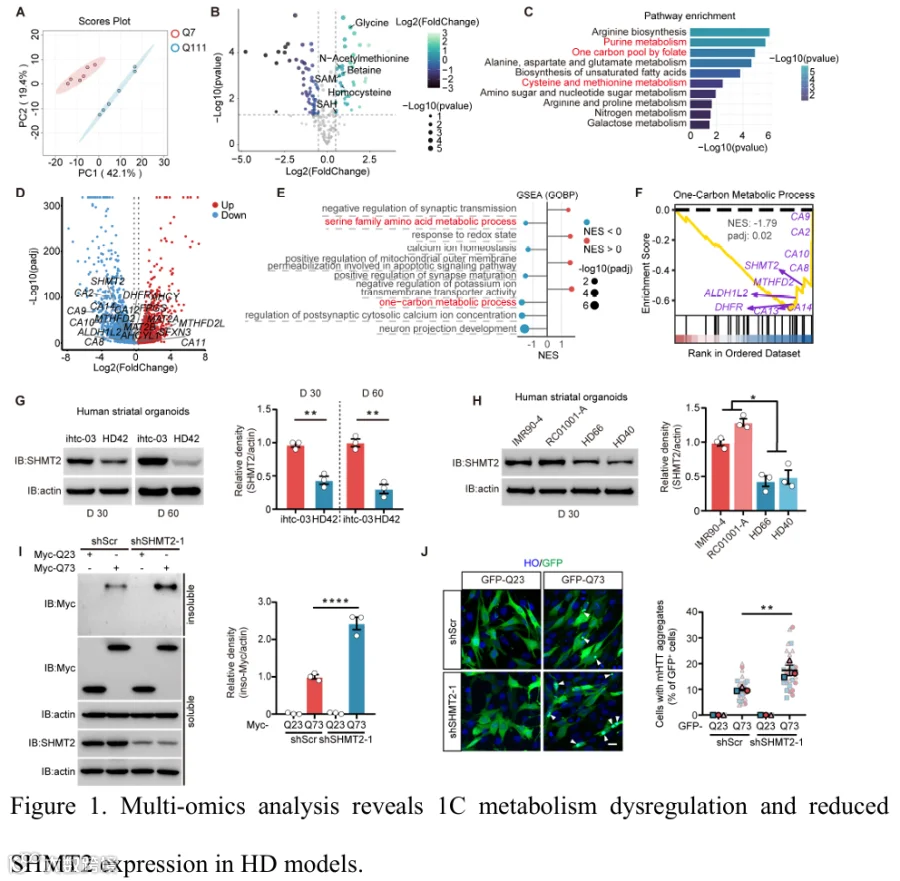
2.SHMT2缺失在iPSC来源的hSO中诱导神经元变性
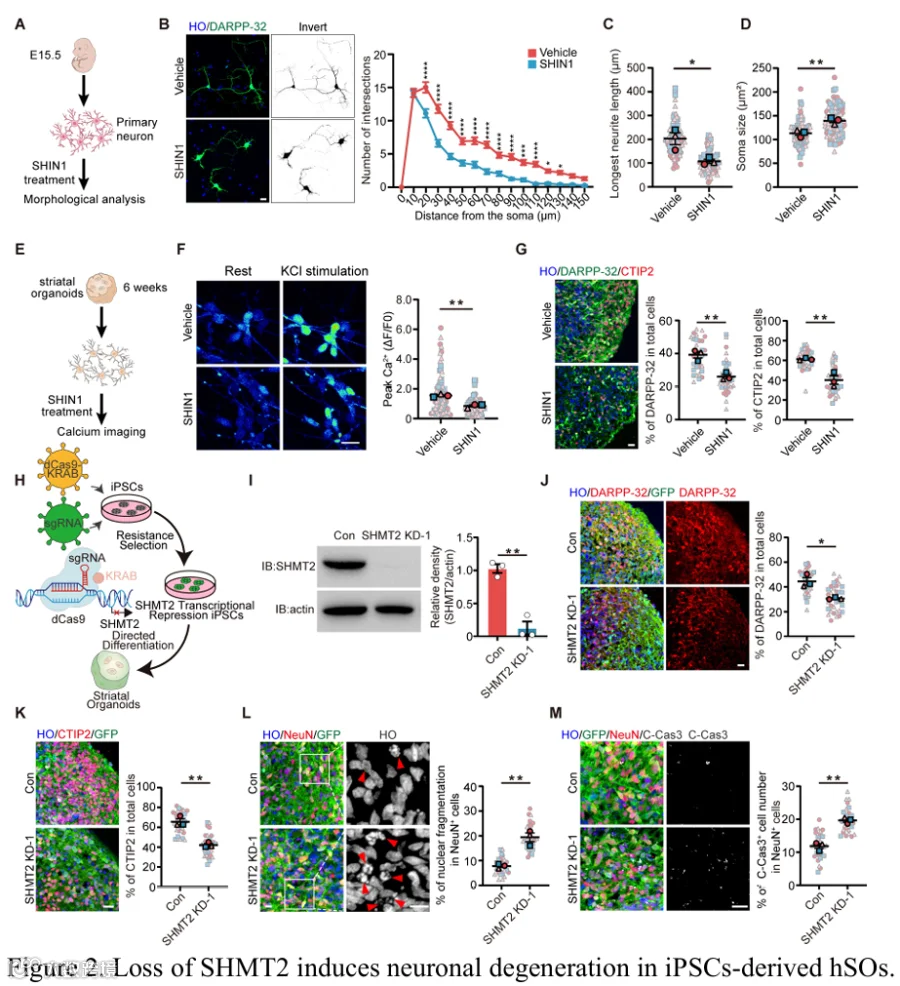
为了评估SHMT2在神经元中的功能相关性,我们首先在HdhQ7和HdhQ111细胞中使用SHIN1(一种SHMT1/2特异性抑制剂)进行了药理学抑制。免疫染色显示,SHIN1处理导致原代纹状体神经元中SHMT2水平降低,并显著损害神经突复杂性,表现为神经突交叉点减少、最长神经突长度缩短和胞体面积增加(图2A-D)。为了评估神经元功能,我们测量了hSO衍生神经元中的钙信号,发现SHMT抑制显著降低了KCl刺激引起的峰值Ca²⁺反应(图2E和2F)。此外,我们分析了DARPP-32⁺和CTIP2⁺神经元群体(MSN的关键标志物),发现SHIN1处理的hSO中这些群体的比例显著降低(图2G),同时DARPP-32⁺神经元中cleaved-caspase-3阳性凋亡增加(补充图2A)。我们接下来使用CRISPR干扰技术生成了SHMT2表达降低的iPSC系(图2H)。使用两个独立的sgRNA,SHMT2敲低降低了hSO中DARPP-32⁺ MSN和CTIP2⁺神经元的比例,单细胞分析确认了DARPP-32⁺ MSN群体的特异性减少,并且通过Sholl分析评估的神经突复杂性也受损(图2I-K和补充图2B-F)。重要的是,SHMT2缺失导致显著的神经元凋亡,在hSO的NeuN⁺神经元和分离的DARPP-32⁺神经元中观察到cleaved caspase-3升高和核碎片化(图2L和2M,以及补充图2G-I)。总之,这些结果证明SHMT2对于在hSO中维持MSN结构完整性、功能活性和存活至关重要。
3.SHMT2缺陷在体内诱导神经变性和运动功能障碍
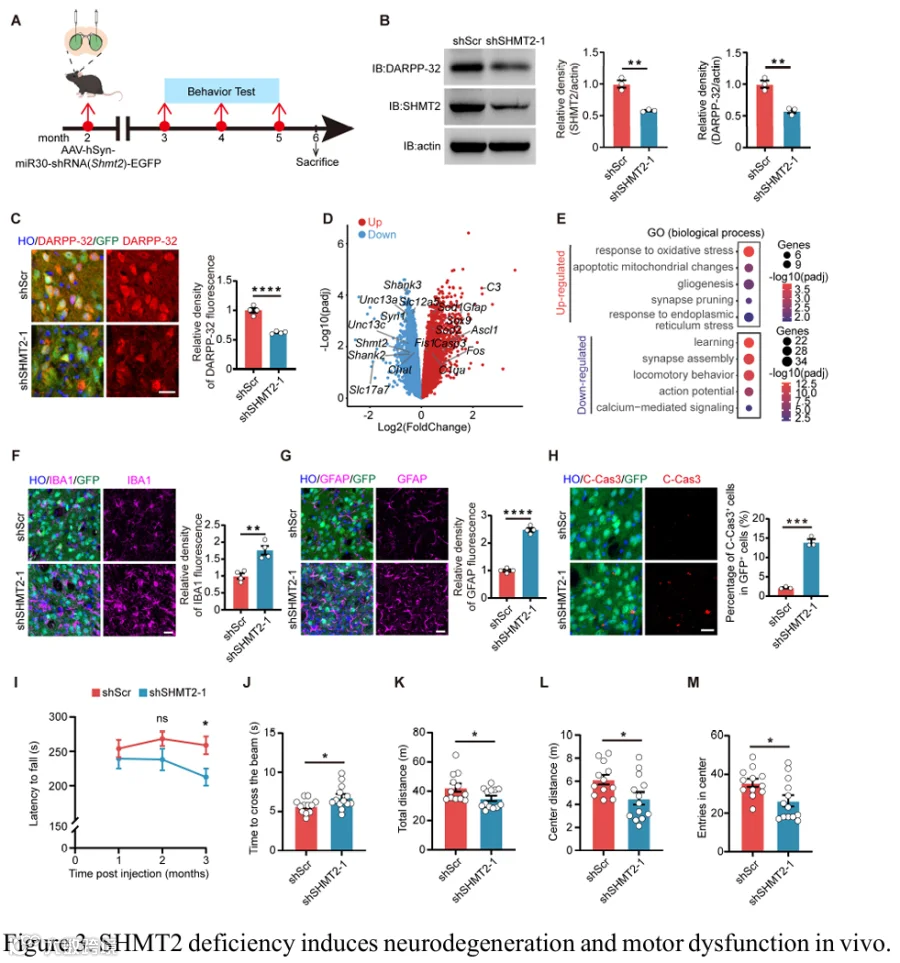
为了进一步研究SHMT2在体内的作用,我们生成了携带靶向SHMT2的shRNA的AAV载体。2月龄C57BL/6小鼠接受纹状体立体定位注射AAV-shSHMT2或对照AAV-shScr,随后进行行为测试和生化分析(图3A)。在AAV-shSHMT2注射小鼠的纹状体中,SHMT2敲低导致DARPP-32显著下调(图3B和3C,以及补充图3A和3B)。为了描绘SHMT2敲低诱导的转录变化,我们对SHMT2敲低小鼠及其对照同窝小鼠的纹状体进行了比较RNA-seq。GO分析显示,与氧化应激和线粒体损伤相关的通路显著激活,同时伴随与突触组织、动作电位发放和钙介导信号相关的神经元功能程序受抑(图3D和3E,补充图3C,补充表4)。同时,GFAP⁺星形胶质细胞和IBA1⁺小胶质细胞的荧光密度明显增加,提示反应性胶质增生和进行性神经退行过程(图3F和3G,以及补充图3D和3E)。此外,SHMT2敲低显著增加了GFP⁺ AAV感染区域内cleaved caspase-3⁺细胞的数量,反映了神经元凋亡增强(图3H和补充图3F)。为了评估运动功能,我们对SHMT2敲低小鼠进行了行为测试。在转棒测试中,注射AAV-shSHMT2的小鼠表现出掉落潜伏期显著缩短,表明运动协调和平衡受损(图3I和补充图3G)。类似地,平衡木行走表现也受损,SHMT2敲低小鼠相比对照需要更多时间穿过平衡木(图3J和补充图3H)。在旷场测试中,SHMT2敲低小鼠显示总移动距离减少、中心区域进入次数减少和中心区域距离减少,反映了运动和探索行为受损(图3K-M和补充图3I-K)。总之,这些发现揭示体内SHMT2敲低会损害MSN完整性,诱导反应性胶质增生和神经元凋亡,并最终导致运动功能障碍。
4.SHMT2过表达在体内外改善神经变性
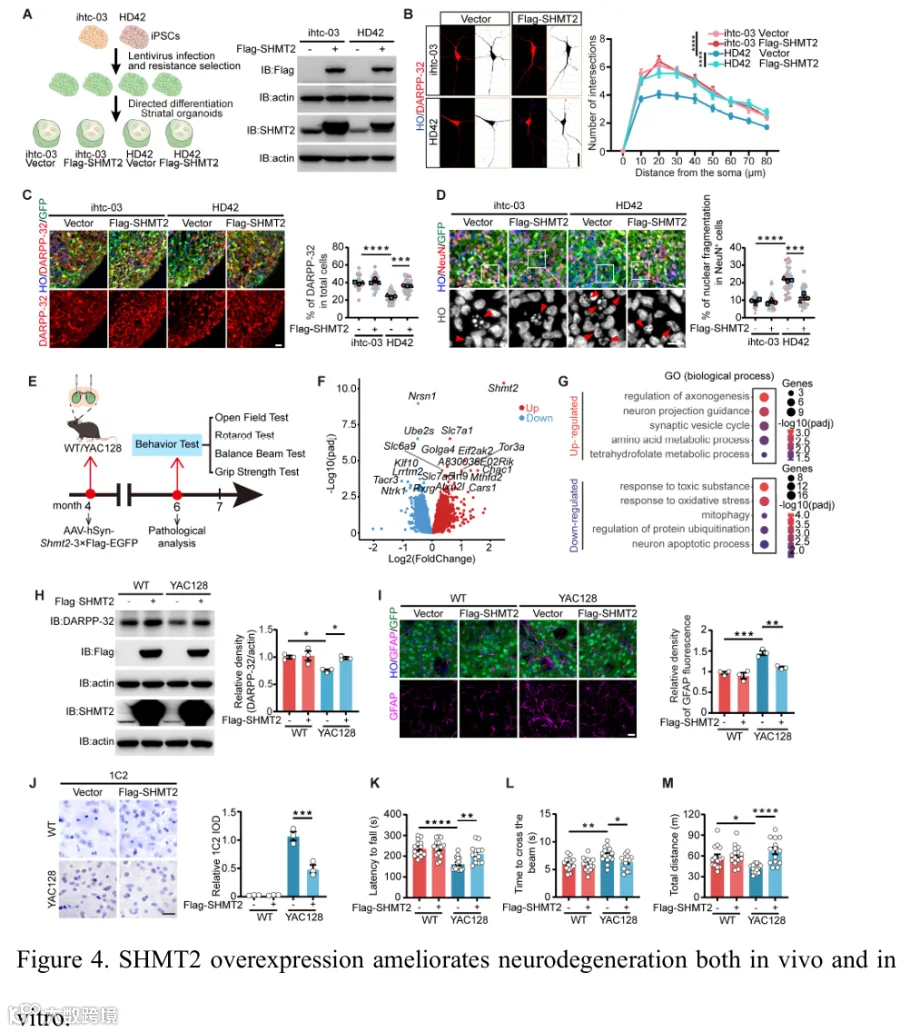
为了探索SHMT2对HD相关病理的影响,我们首先将慢病毒介导的SHMT2过表达导入对照和HD患者来源的iPSC中,随后筛选SHMT2表达细胞并分化成hSO(图4A)。在HD-hSO中,我们观察到神经突生长减少和结构异常,表明神经元完整性受损(图4B)。值得注意的是,SHMT2过表达增强了神经突长度和分支,提示在维持神经元结构和稳定性中起保护作用(图4B和补充图4A)。免疫染色分析显示,SHMT2过表达增加了HD-hSO中DARPP-32⁺和CTIP2⁺神经元的比例(图4C和补充图4B-E)。同时,它显著减少了显示核碎片化的细胞数量(图4D和补充图4F-G),表明SHMT2促进神经元身份并减轻HD相关的细胞变性。为了确定这些细胞改善是否转化为体内功能益处,我们接下来在YAC128 HD小鼠中评估了AAV-SHMT2递送后的分子和行为结果(图4E)。转录组分析显示,SHMT2过表达部分逆转了YAC128相关的树突发育、突触组织和线粒体应激反应通路的缺陷(图4F和4G,补充图4H,补充表5)。蛋白质印迹分析显示,SHMT2过表达后纹状体中的DARPP-32水平恢复,表明MSN完整性改善(图4H)。SHMT2过表达还减轻了YAC128小鼠GFP⁺纹状体区域内的星形胶质细胞反应性,并降低了mHTT免疫反应性和不溶性mHTT水平,与HD相关病理特征的更广泛改善一致(图4I和4J,以及补充图4I)。功能上,AAV-SHMT2处理的YAC128小鼠表现出显著改善的运动表现。在转棒测试中,小鼠表现出显著延长的掉落潜伏期,反映了协调性增强(图4K)。平衡木分析显示更短的穿越时间,表明平衡改善(图4L)。握力也相比对照小鼠明显改善(补充图4J)。最后,在旷场测试中,处理动物移动总距离更长,在中心区域移动更频繁,并做出更多中心区域进入(图4M和补充图4K-L)。总之,这些发现将SHMT2鉴定为HD中神经元结构、功能和存活的关键调节因子,能够对抗MSN退变并改善行为缺陷。
5.SHMT2缺陷通过HCY积累驱动神经变性
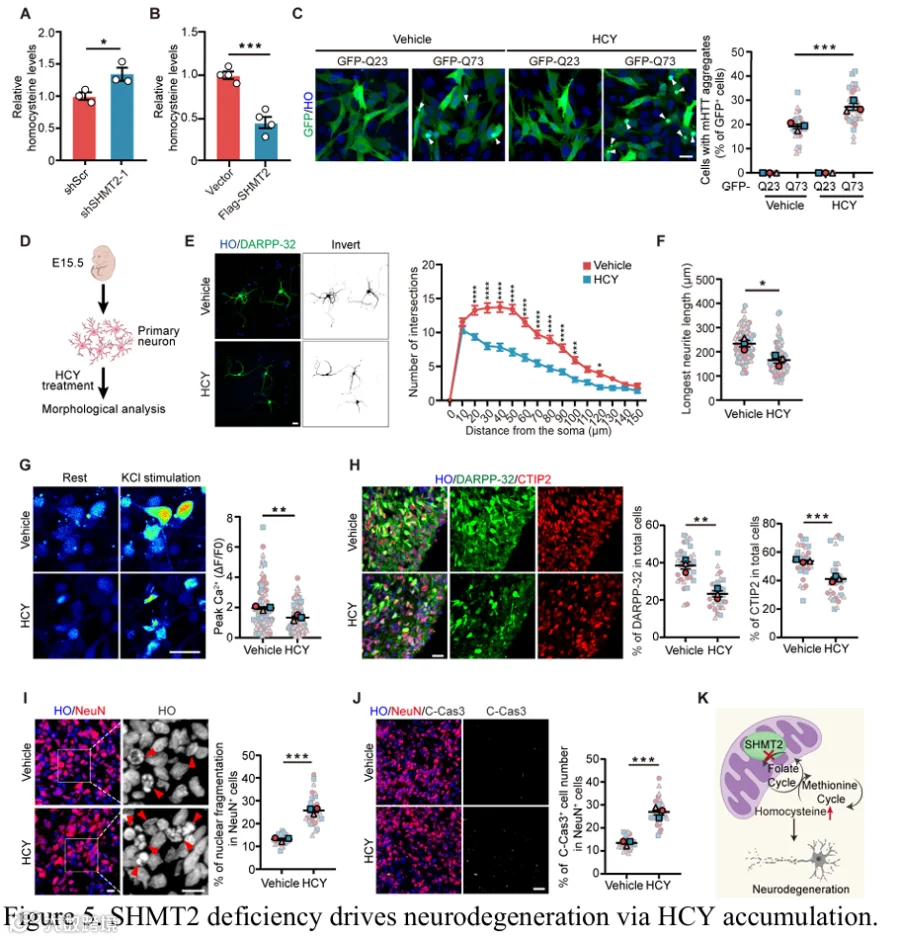
HCY是一碳代谢途径中的关键中间代谢物,已被证明与神经毒性和神经退行性变有关。靶向代谢组学分析显示,与对照相比,HD模型中HCY水平显著升高(补充图1A)。鉴于SHMT2是mt-1C代谢的核心调节因子,我们假设HD中SHMT2缺陷可能破坏1C代谢稳态,导致HCY积累。支持这一假设的是,我们发现SHMT2敲低导致细胞内HCY水平升高(图5A),而HdhQ111细胞中SHMT2过表达有效降低了HCY浓度(图5B)。为了检查HCY对HD相关病理的影响,我们首先研究了HCY是否调节mHTT聚集。使用过表达Q73的HdhQ7细胞模型,我们发现HCY处理显著增加了具有polyQ聚集体的细胞数量(图5C),提示升高的HCY水平可能加剧HD中的蛋白毒性应激。为了进一步评估HCY对神经元完整性的影响,我们用HCY处理原代MSN,观察到明显的形态学改变,包括神经突复杂性降低和最長神经突显著缩短(图5D-F),提示神经元萎缩和早期退变。细胞内钙成像显示,HCY处理的hSO衍生神经元对KCl诱导的钙内流反应减弱,反映神经元兴奋性和突触信号受损(图5G)。此外,HCY暴露导致DARPP-32⁺和CTIP2⁺神经元群体减少,表明MSN丢失和纹状体神经元身份破坏(图5H)。此外,HCY处理增加了hSO中NeuN阳性神经元内的核碎片化并升高了cleaved caspase-3水平,证实了凋亡活性增强和神经元死亡(图5I和5J)。总之,这些发现表明SHMT2缺陷导致HCY积累,后者加剧polyQ聚集,损害神经元结构和功能,并通过凋亡途径促进神经变性(图5K)。
6.HCY通过AARS1抑制组蛋白乳酸化
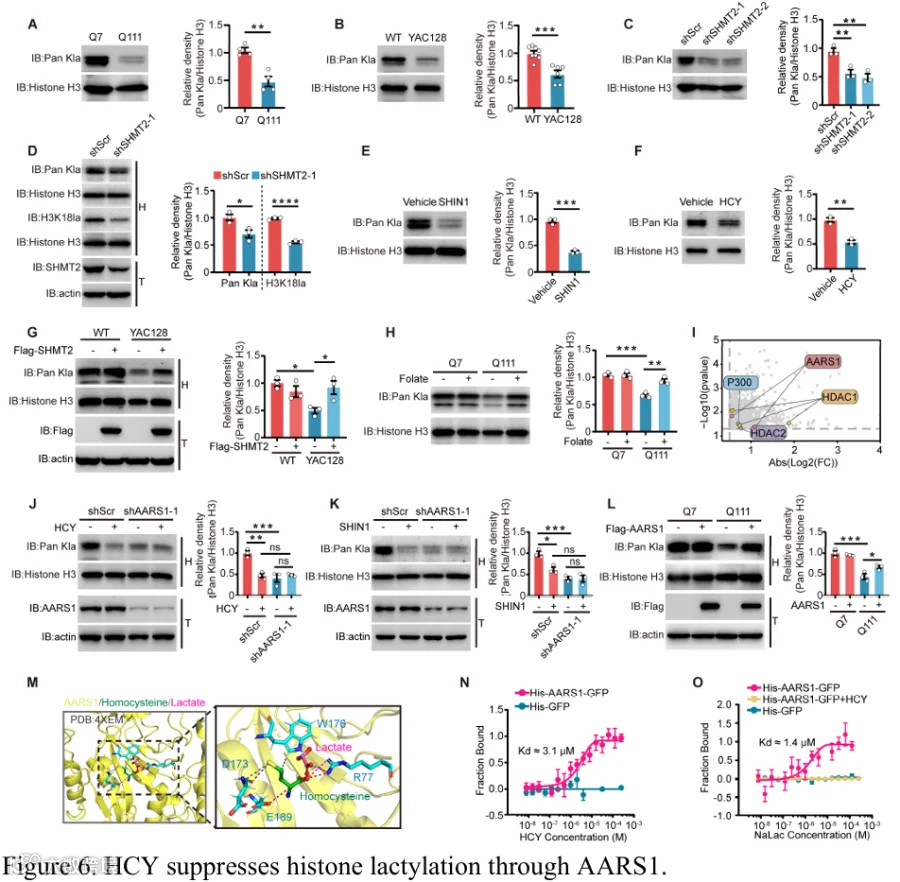
组蛋白乳酸化是一种新兴的翻译后修饰,将代谢状态与转录调控联系起来,在神经元功能和稳态中发挥关键作用。代谢中间产物可通过影响相关酶的活性来调节组蛋白修饰。在此背景下,我们推测由1C代谢受损导致的HCY水平升高可能促成组蛋白乳酸化的失调。为了验证这一点,我们首先评估了HdhQ7和HdhQ111纹状体细胞以及WT和YAC128小鼠纹状体中的乳酸化水平。与对照相比,HdhQ111细胞和YAC128小鼠纹状体均表现出组蛋白乳酸化显著降低(图6A和6B),表明HD模型中存在一致的表观遗传破坏。SHMT2敲低或使用SHIN1的药理学抑制产生了类似的组蛋白乳酸化降低(图6C-E),提示SHMT2缺陷是一个促成因素。值得注意的是,位点特异性乳酸化分析显示HdhQ111细胞中广泛下降,SHIN1处理的细胞中也密切反映(补充图5A和5B)。HCY处理进一步抑制了组蛋白乳酸化(图6F),加强了HCY积累与表观遗传扰动之间的联系。重要的是,SHMT2过表达恢复了HD模型中的乳酸化水平(图6G和补充图5C和5D)。作为补充,叶酸补充通过增强叶酸循环活性和减少HCY积累,也挽救了HD模型和SHMT2缺陷条件下的组蛋白乳酸化缺陷(图6H和补充图5E),强调了HCY稳态在调节该表观遗传修饰中的关键作用。为了进一步阐明HCY影响组蛋白乳酸化水平的机制,我们进行了有限蛋白水解-质谱分析(补充图5F),鉴定出1,309个显著改变的肽段(补充表6),对应几个潜在的HCY相互作用蛋白,包括AARS1、p300、HDAC1和HDAC2——所有这些先前均已被证明参与组蛋白乳酸化调节(图6I)。为了验证它们在纹状体细胞中的功能相关性,我们单独敲低了每个候选基因。值得注意的是,沉默p300、HDAC1或HDAC2对组蛋白乳酸化影响甚微,而AARS1敲低显著抑制了全局组蛋白乳酸化水平,表明AARS1在此背景下具有特定作用(图6J和补充图5G和5H)。此外,AARS1缺失消除了HCY诱导的组蛋白乳酸化降低(图6J和补充图5I),表明AARS1介导HCY积累的表观遗传效应。此外,AARS1敲低阻止了由SHMT2敲低或SHIN1处理引起的组蛋白乳酸化降低(图6K和补充图5J和5K)。相反,在HdhQ111细胞中过表达AARS1导致组蛋白乳酸化水平显著增加(图6L)。有趣的是,尽管有这些功能效应,AARS1蛋白水平在多个HD模型中保持不变(补充图5L-N),提示HCY可能通过调节AARS1活性而非改变其表达来调节组蛋白乳酸化。为了进一步探索HCY调节AARS1活性的机制,我们对HCY和乳酸与AARS1的晶体结构进行了分子对接分析。结果显示HCY和乳酸在R77残基处存在空间重叠,提示HCY可能在AARS1活性位点取代乳酸以发挥其功能效应的潜在竞争性结合机制(图6M)。微量热泳动分析显示HCY可直接结合AARS1并竞争性干扰乳酸结合,提供了HCY通过竞争性结合破坏AARS1介导的组蛋白乳酸化的直接证据(图6N和6O,以及补充图5O)。总之,这些结果表明升高的HCY竞争性破坏AARS1介导的组蛋白乳酸化,促成了HD模型中观察到的表观遗传失调。
7.氟哌啶醇通过SHMT2依赖性机制减轻代谢-表观遗传失调
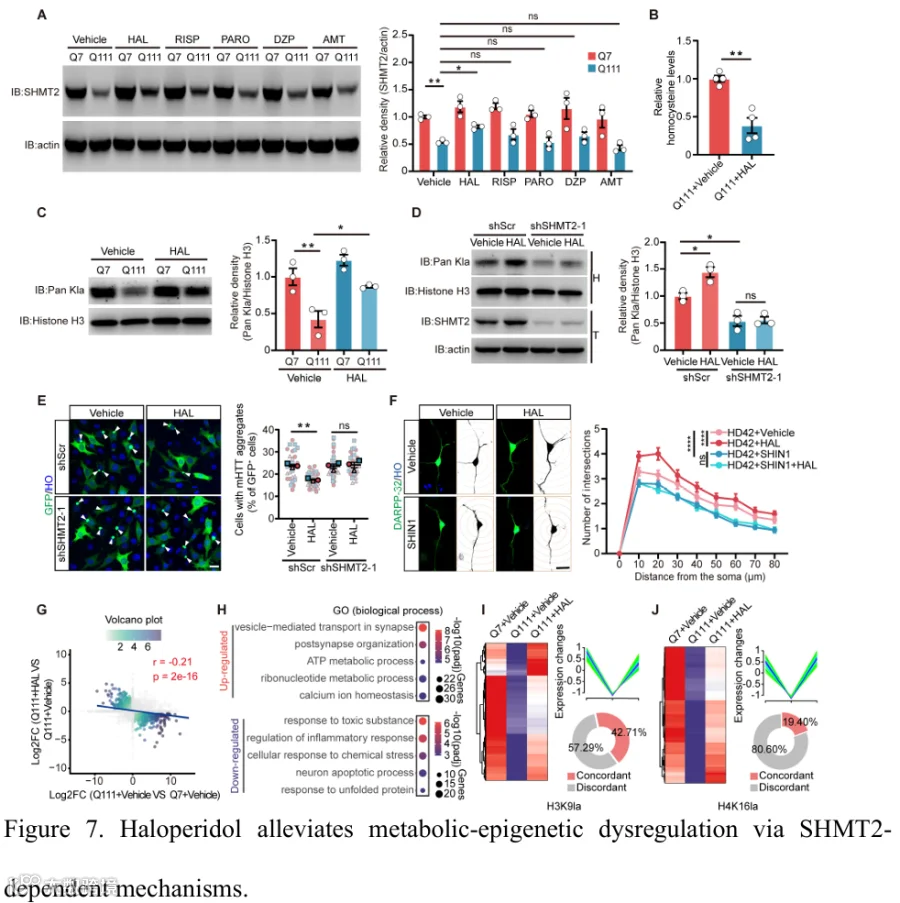
为了确定与HD相关的对症药物是否能在体外调节SHMT2调节的组蛋白乳酸化,我们筛选了一系列药理学药物,包括氟哌啶醇、利培酮、金刚烷胺、帕罗西汀和多奈哌齐,这些药物通常用于HD的对症管理。在这些药物中,只有氟哌啶醇在HdhQ111细胞中显著上调SHMT2表达,而其他药物没有表现出显著效果(图7A)。然而,氟哌啶醇处理并未显著改变HdhQ7和HdhQ111细胞中Shmt2 mRNA水平,提示氟哌啶醇可能在蛋白水平调节SHMT2(补充图6A)。此外,与未处理对照相比,氟哌啶醇处理降低了HdhQ111细胞中的细胞内HCY水平,并恢复了在HdhQ111细胞中降低的组蛋白乳酸化水平(图7B和7C)。重要的是,通过遗传敲低抑制SHMT2消除了氟哌啶醇诱导的组蛋白乳酸化增加(图7D),表明氟哌啶醇通过调节SHMT2-HCY轴减轻HD模型中的代谢-表观遗传失调。免疫荧光染色显示,氟哌啶醇处理明显减少了携带polyQ聚集体的细胞数量,而这种效应被SHMT2敲低所消除(图7E和补充图6B)。一致地,使用SHIN1对SHMT2的药理学抑制也阻断了氟哌啶醇介导的神经元形态恢复(图7F和补充图6C-E),表明这些效应需要SHMT2活性。鉴于氟哌啶醇恢复组蛋白乳酸化,后者与转录调控密切相关,我们接下来检查其效应是否通过组蛋白乳酸化依赖的基因转录变化介导。为此,我们用氟哌啶醇处理HdhQ111细胞,并对Q7、Q111和Q111加氟哌啶醇组进行RNA测序(补充图6F和补充表7)。比较Q111 vs. Q7和Q111+HAL vs. Q111的双火山图揭示了一组响应氟哌啶醇表现出逆转表达模式的基因,两个比较之间呈负相关(r=-0.21),支持其部分恢复转录稳态(图7G)。重叠基因的热图可视化进一步支持了这一观察,显示许多受影响的基因在Q111相对于Q7中下调,随后在氟哌啶醇处理后上调,反之亦然——表明基因水平的挽救效应(补充图6G)。在Q111+HAL vs. Q111比较中鉴定出的4,012个差异表达基因中,有1,312个与Q111 vs. Q7组的DEGs重叠并表现出相反的表达趋势。这些基因被定义为氟哌啶醇挽救的靶点,约占HAL调节转录组的33%(补充图6H)。上调和下调基因集的GO分析鉴定了与突触组织、代谢过程、钙离子稳态、炎症和应激反应以及神经元凋亡相关的通路(图7H)。CUT&Tag-RNA-seq联合分析显示,由H3K9la或H4K16la标记的基因中有相当一部分在氟哌啶醇处理后表现出一致的表达变化,表明氟哌啶醇诱导的转录反应与其对组蛋白乳酸化的效应部分一致(图7I和7J,以及补充表8)。这些一致基因的GO和通路富集分析进一步揭示了与神经元组织、突触和囊泡相关过程以及细胞代谢调节相关的功能类别富集(补充图6I和6J)。总之,这些发现表明氟哌啶醇在HD模型中部分恢复了SHMT2依赖的代谢和表观遗传稳态。
更多结果和补充图表:doi:10.1172/JCI196094


扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:
https://cnsknowall.com
CNSknowall:24年最新问世的遥遥领先的科研数据(0代码生信+统计学)分析平台,同时含有机制图模块+汉化版Pubmed融合Deepseek高效筛选目标文献+SCI文献例句/语料检索模块+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动批阅!