今天给大家解读一篇3月发表在《Frontiers in Cell and Developmental Biology》上的题目为“PRMT5 inhibition triggers functional ATM deficiency and sensitizes pancreatic cancer to CHK1 blockade.”的文章。该研究聚焦PRMT5抑制剂在PDAC中的治疗潜力。作者通过体外和体内实验发现,PRMT5抑制可导致ATM蛋白显著下降,使细胞处于功能性ATM缺失状态,进而依赖ATR-CHK1通路维持存活。联合抑制PRMT5和CHK1能有效协同杀伤PDAC细胞,并在小鼠模型中实现显著抑瘤效果。文章从分子、细胞、转录组和动物整体水平验证了这一组合策略的合理性和有效性。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!包括数据集、绘图代码、图表复现、思路总结、参考文献……0代码!鼠标点点点即可轻松完成5-10分生信SCI全文复现!
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!









团队成员合影(位于上海陆家嘴中心,可随时预约参观)






题目:《PRMT5抑制会引发功能性的ATM缺陷并使胰腺癌对CHK1阻断敏感》PRMT5 inhibition triggers functional ATM deficiency and sensitizes pancreatic cancer to CHK1 blockade
发表期刊:Frontiers in Cell and Developmental Biology
影响因子:4.3
研究背景:
PRMT5抑制剂正处于临床研究阶段用于治疗PDAC,但如何最大化其疗效的策略尚不明确。本研究旨在确定药理抑制PRMT5是否会在PDAC中产生可被治疗利用的脆弱性。
研究思路:
- 分子与细胞实验
采用免疫荧光、Western blotting、彗星实验分析ATM水平及DNA损伤应答;使用IncuCyte活细胞成像和克隆形成实验评估体外生长;通过RNA-seq进行转录组分析,并用RT-qPCR阵列验证选定靶点。 - 体内验证
建立皮下异种移植模型,结合免疫组化检测肿瘤组织中ATM、Ki67和γH2A.X的表达,评估联合治疗对肿瘤体积、中位生存期及体重的影响。
研究亮点:
-
首次阐明PRMT5抑制通过降低ATM水平、诱导功能性ATM缺陷,从而迫使PDAC细胞依赖ATR-CHK1通路生存。 -
验证了PRMT5与CHK1双重抑制在体外(细胞生长抑制、凋亡诱导、DNA损伤积累)和体内(肿瘤体积缩小、生存期延长)均具有协同抗肿瘤效应。 -
通过RNA-seq结合RT-qPCR验证,从转录组水平揭示了联合治疗下调细胞周期和DNA修复基因、上调细胞死亡通路的分子机制。
研究结果:
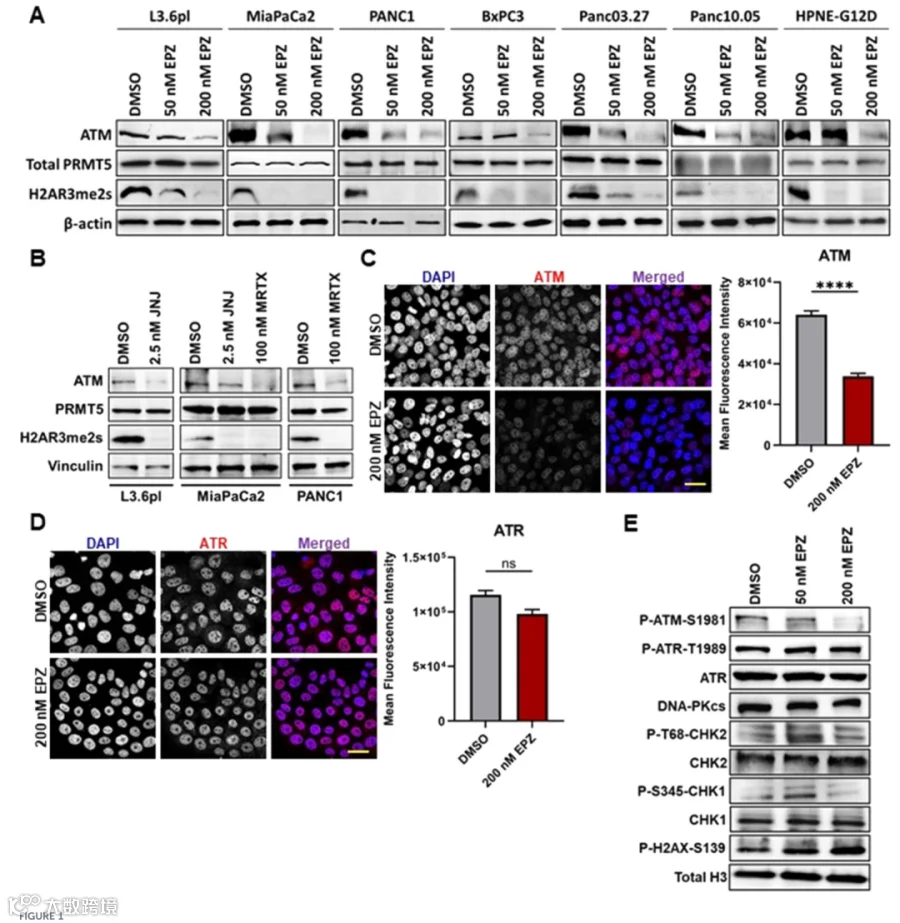
- ATM水平下降
使用不同PRMT5抑制剂处理多种PDAC细胞系,均显著降低ATM蛋白水平,导致功能性ATM缺陷状态。 - 信号通路重塑
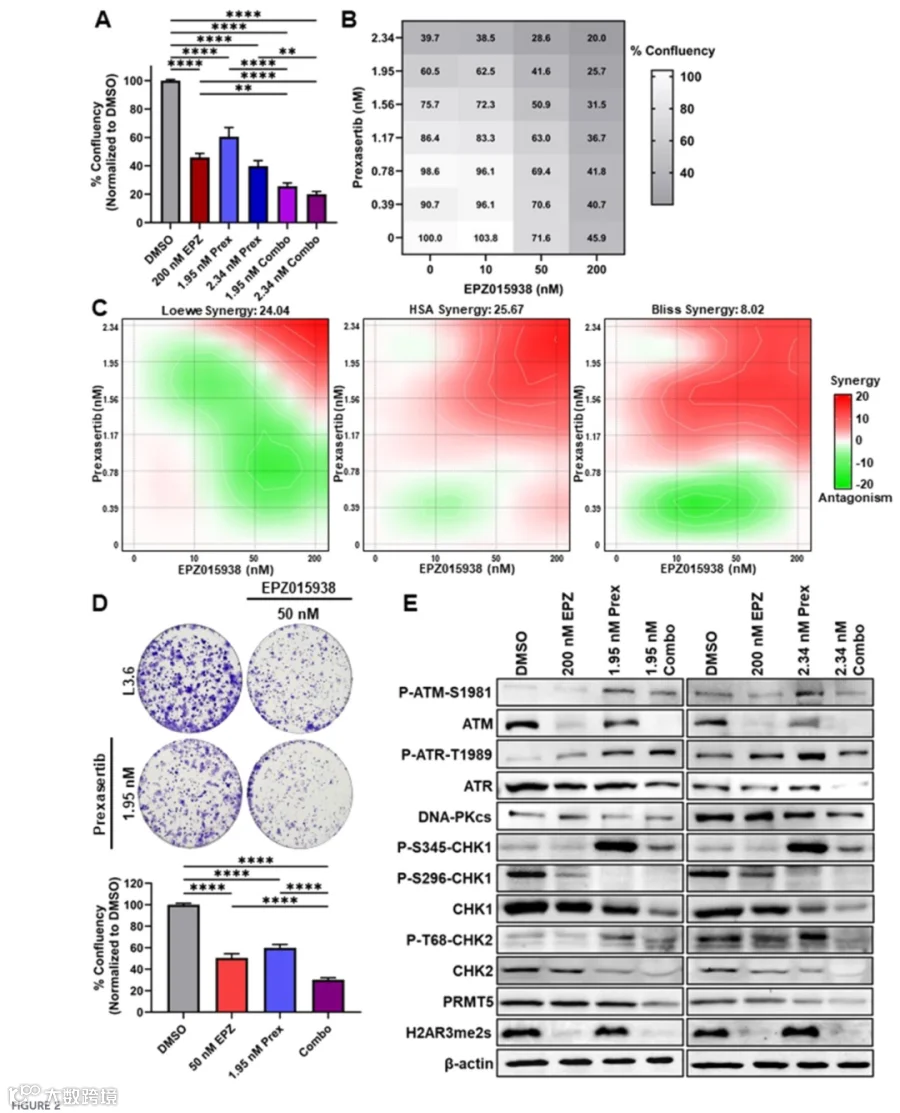
ATM减少重新编程DNA损伤应答信号,PDAC细胞对ATR-CHK1通路的依赖性增强。 - 联合治疗的协同效应
PRMT5与CHK1联合抑制显著抑制PDAC生长,伴随Caspase 3/7活化增强、Annexin V染色阳性率升高及DNA损伤积累增加。 - 转录组变化
RNA-seq显示细胞周期和DNA修复基因下调,细胞死亡通路上调,为协同效应提供机制依据。 - 体内疗效
皮下异种移植模型中,联合治疗显著减少肿瘤体积,延长中位生存期,且未对体重产生不良影响。治疗后的肿瘤组织中ATM和Ki67表达降低,γH2A.X水平升高。
研究总结:
结果译文:
1.PRMT5抑制在PDAC细胞中诱导功能性ATM缺失并重塑DDR信号
2.PRMT5与CHK1联合抑制协同性地抑制PDAC细胞生长与存活
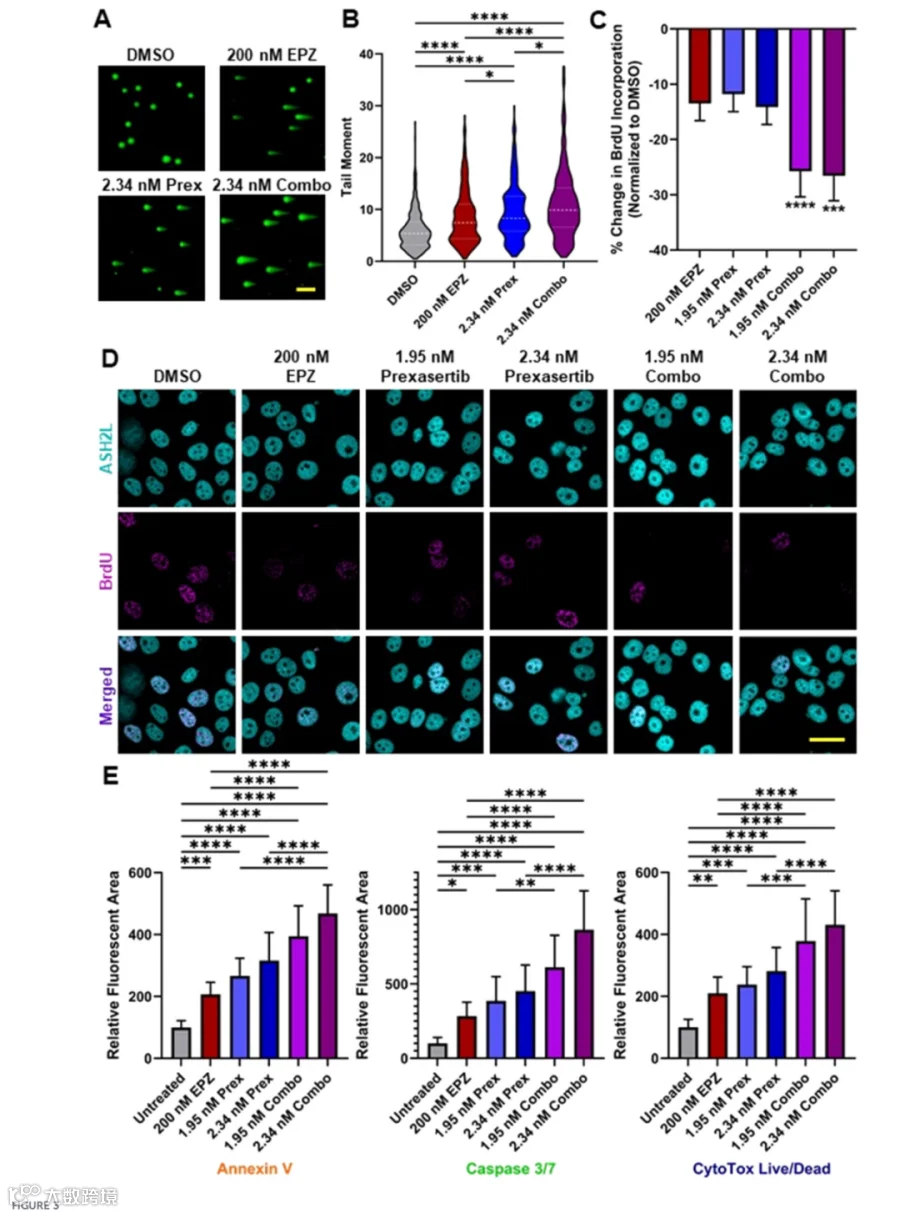
3.PRMT5与CHK1双靶向改变PDAC细胞中调控DNA修复、细胞周期进程和细胞死亡的基因表达网络
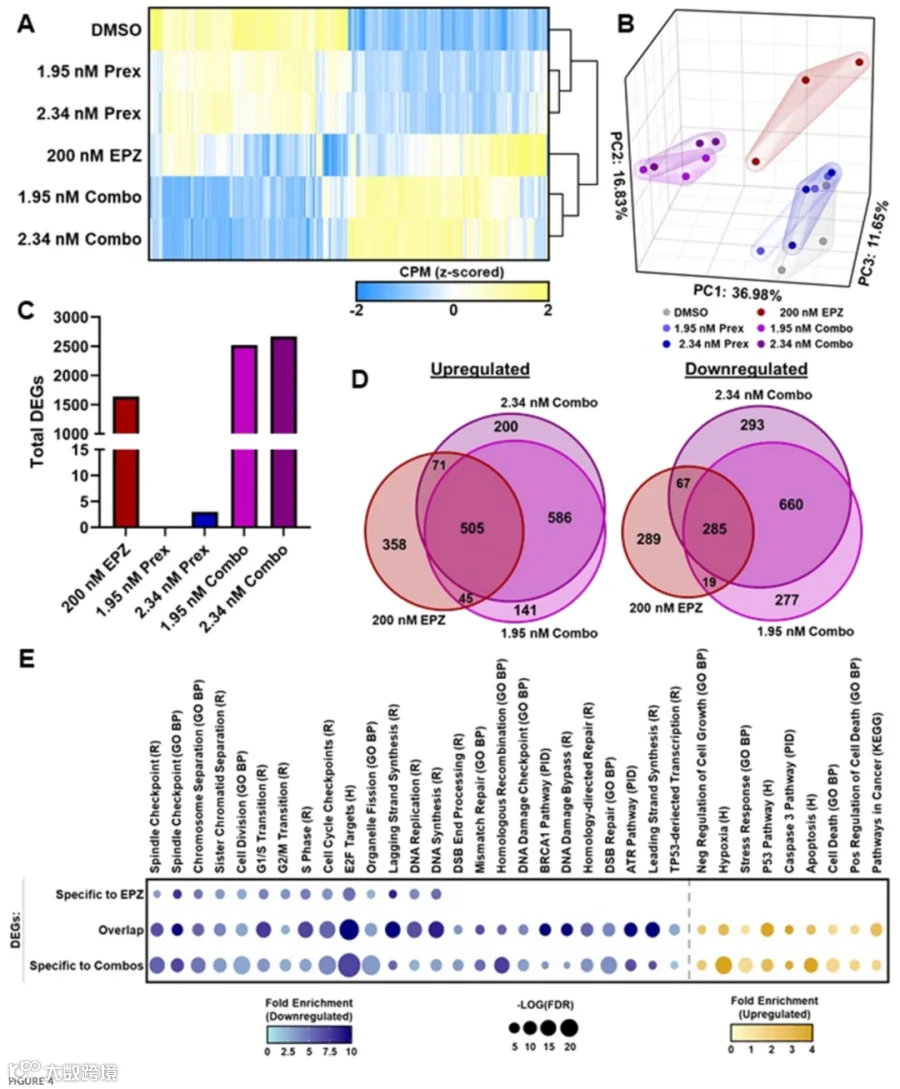
鉴于PRMT5与CHK1联合抑制的体外疗效以及PRMT5已知的表观遗传功能,我们接着研究了L3.6 PL细胞中双抑制的转录后果。在RNA测序之前,细胞经200 nM EPZ015938和1.95–2.34 nM prexasertib单独或联合处理5天。在所有处理组中,共鉴定出3797个差异表达基因(DEGs;相对溶媒对照,倍数变化≥ ±2,FDR < 0.05)(图4A)。DEGs热图可视化显示处理组间存在不同的基因聚类,主成分分析(PCA)进一步证实了这一点。PCA显示prexasertib处理样本与溶媒对照紧密聚类,提示prexasertib单独引发的转录扰动极小(图4B)。相反,EPZ处理和联合处理样本聚在一起,表明存在共同且潜在的协同效应。值得注意的是,总DEGs汇总证实prexasertib单独引发极少的转录改变,>99%的DEGs来自EPZ或联合处理(图4C;补充图S3)。比较分析鉴定出EPZ单独上调的979个基因,而联合处理诱导了额外的927个基因在共处理样本中专属上调(图4D)。类似地,EPZ单独下调660个基因,联合处理额外下调1230个基因(图4D)。联合处理所致的更高水平转录改变,以及prexasertib单独的微弱效应,提示PRMT5抑制建立了一种许可性的表观遗传景观,使基因网络对CHK1通路扰乱更为响应。为进行功能解读,我们使用MSigDB进行通路富集分析,并将结果可视化为气泡图以描绘通路显著性和标准化富集分数(图4E)。基因被归类为EPZ专属、联合处理专属或两者共享。PRMT5抑制单独显著重塑了转录组,而与CHK1共抑制则增强了这些效应并激活了独特、额外的转录程序。如图4D所示,与EPZ单独相比,联合处理细胞呈现更多数量的上调和下调基因。相应地,通路富集分析(图4E;补充表S2)揭示了共享通路的扩增及新通路的参与,联合处理一致性地影响了重叠网络中额外的基因。富集通路反映了早期实验所观察到的生物学表型,包括细胞周期进程和DNA复制/修复基因的下调,以及细胞死亡相关过程的上调(图4E;补充表S2)。为验证这些发现,我们进行了靶向DNA损伤信号通路基因的RT-qPCR阵列,结果证实了RNA-seq模式(补充图S4;补充表S3,S4)。总而言之,这些分子特征证明,虽然PRMT5抑制单独可抑制增殖和修复相关转录程序,但PRMT5与CHK1联合抑制驱动了更广泛且更显著的转录重编程,增强DNA损伤积累,强制执行细胞周期阻滞,并促进细胞死亡。
4.PRMT5与CHK1联合抑制增强PDAC异种移植模型的抗肿瘤疗效并延长生存期
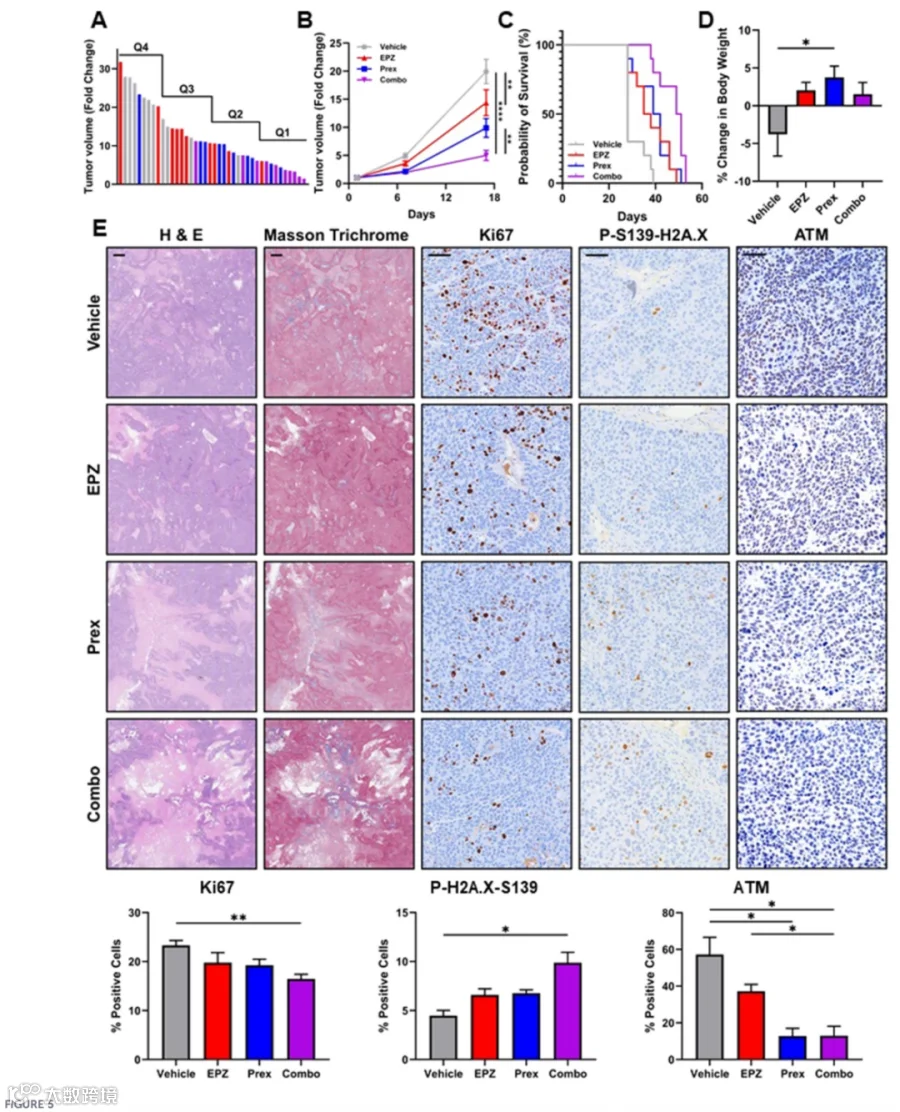
此后,我们利用皮下L3.6 PL异种移植模型评价了PRMT5与CHK1联合抑制的体内治疗疗效。肿瘤达100 mm³后,将小鼠随机分为四组,分别接受溶媒、EPZ015938、prexasertib或联合用药。肿瘤体积FC分布分析显示,大多数联合治疗肿瘤(90%)落入两个最低四分位数内,其中70%处于最低四分位数,对应于最小的肿瘤体积(图5A)。单因素方差分析显示治疗对第17天肿瘤生长的总体效应显著(p < 0.0001)。事后比较揭示,与对照组相比,EPZ015938显著降低肿瘤生长(p ≤ 0.01;9.8±1.6 FC vs. 19.9±2.1 FC),而prexasertib单独则未达显著(p > 0.05;14.4±2.2 FC vs. 19.9±2.1 FC)。联合处理产生的效应最强,使肿瘤生长降低至5.0±0.9 FC(p ≤ 0.0001 vs. 对照),且显示优于任一单独治疗的抑制作用(p ≤ 0.05 vs. prexasertib;p ≤ 0.01 vs. EPZ015938;图5B)。此外,联合治疗动物的生存期显著延长,中位生存期为50天,而对照组为28天,EPZ015938单独为36.5天,prexasertib单独为40.5天(p ≤ 0.001;图5C)。所有治疗组在整个研究期间体重均保持在基线5%以内。接受治疗的小鼠呈现轻微体重增加(1.5%–3.8%),而未处理对照组平均体重下降3.8%(图5D),表明联合处理并未对体重产生不良影响。切除肿瘤的组织病理学分析显示,联合治疗小鼠的总体肿瘤尺寸缩小、坏死增加,同时Masson三色切片上增强的蓝色纤维染色显示致密的瘤周纤维化。免疫组化分析显示,联合治疗组肿瘤中增殖标志物Ki67显著降低(16.46%±0.96% vs. 对照23.32%±0.99%,p ≤ 0.01)。DNA损伤标志物γH2A.X(P-S139-H2A.X)升高(9.88%±1.05% vs. 对照4.478%±0.54%,p ≤ 0.05),而总ATM染色明显减少(12.98%±5.14% vs. 对照57.26%±9.36%,p ≤ 0.05;图5E)。这些体内发现与我们的体外结果相呼应,证明PRMT5与CHK1联合抑制在PDAC细胞中放大了DNA损伤、抑制了增殖并降低了ATM水平。综合来看,这些数据确立了PRMT5与CHK1双重抑制是一种有前景、耐受性良好的治疗策略,能够在体内限制肿瘤生长并延长生存期。
更多结果和补充图表:doi: 10.3389/fcell.2026.1748541


扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:
https://cnsknowall.com
CNSknowall:24年最新问世的遥遥领先的科研数据(0代码生信+统计学)分析平台,同时含有机制图模块+汉化版Pubmed融合Deepseek高效筛选目标文献+SCI文献例句/语料检索模块+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动批阅!