今天给大家解读一篇4月发表在《Emerging Microbes & Infections》上的题目为“Rapid and precise amplicon-based genome sequencing to preparedness and response chikungunya virus outbreaks.”的文章。该研究旨在确定用于CHIKV基因组监测的最佳测序框架。作者系统评估了四种NGS配置(2nd-seq-200bp, 2nd-seq-400bp, 3rd-seq-200bp, 3rd-seq-400bp),使用13份临床样本(覆盖从强阳性到弱阳性)。研究对比了测序深度、覆盖率、扩增偏差、变异检测能力(特别是低频变异)以及系统发育分型一致性。结果显示,第三代测序平台在均匀覆盖、低频变异恢复和低病毒载量样本性能上占优,而第二代测序平台在测序深度和产量上占优。两种平台在共识基因组分型和系统发育分析上结果一致。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!包括数据集、绘图代码、图表复现、思路总结、参考文献……0代码!鼠标点点点即可轻松完成5-10分生信SCI全文复现!
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!









团队成员合影(位于上海陆家嘴中心,可随时预约参观)






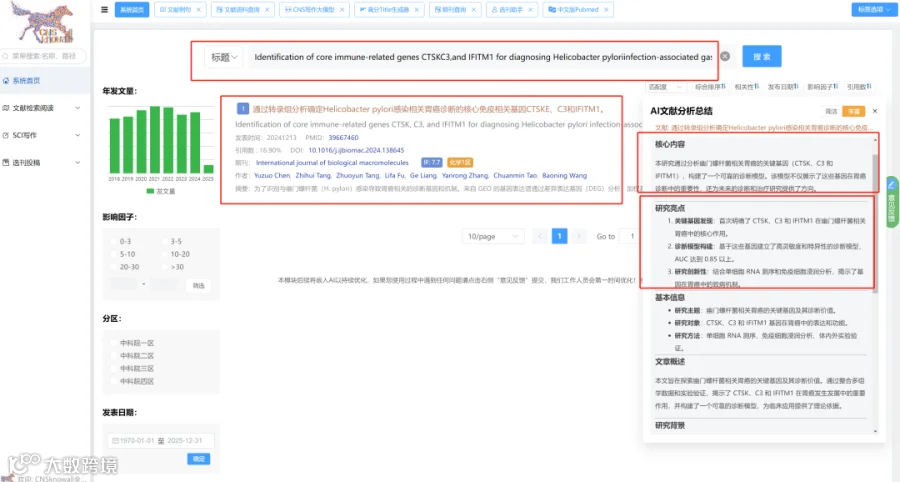
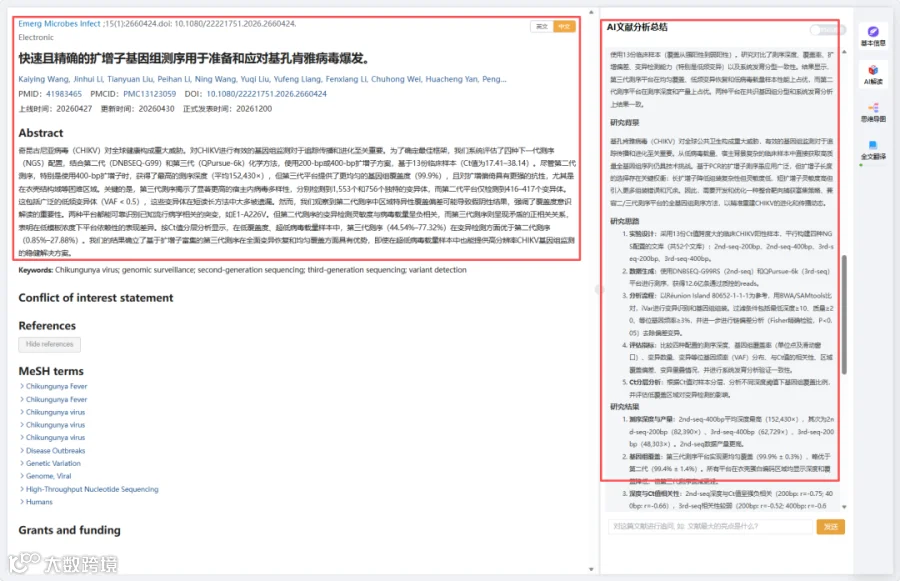
题目:《快速且精确的扩增子基因组测序用于准备和应对基孔肯雅病毒爆发》Rapid and precise amplicon-based genome sequencing to preparedness and response chikungunya virus outbreaks
发表期刊:Emerging Microbes & Infections
影响因子:7.5
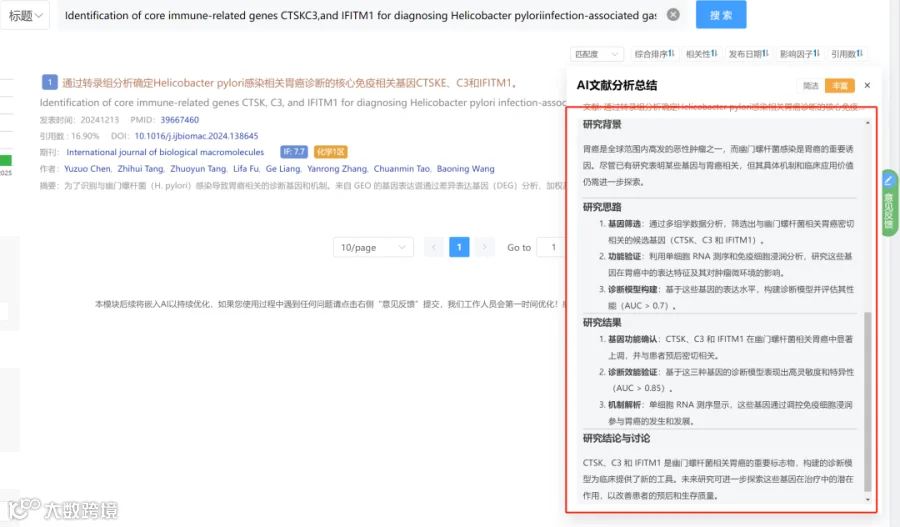
研究背景:
基孔肯雅病毒(CHIKV)对全球公共卫生构成重大威胁,有效的基因组监测对于追踪传播和进化至关重要。从低病毒载量、宿主背景复杂的临床样本中直接获取高质量全基因组序列仍具技术挑战。基于PCR的扩增子测序虽应用广泛,但扩增子长度的选择存在关键权衡:长扩增子降低组装复杂性但灵敏度低,短扩增子灵敏度高但引入更多组装错误和冗余。因此,需要开发和优化一种整合靶向捕获富集策略、兼容二/三代测序平台的全基因组测序方法,以精准重建CHIKV的进化和传播动态。
研究思路:
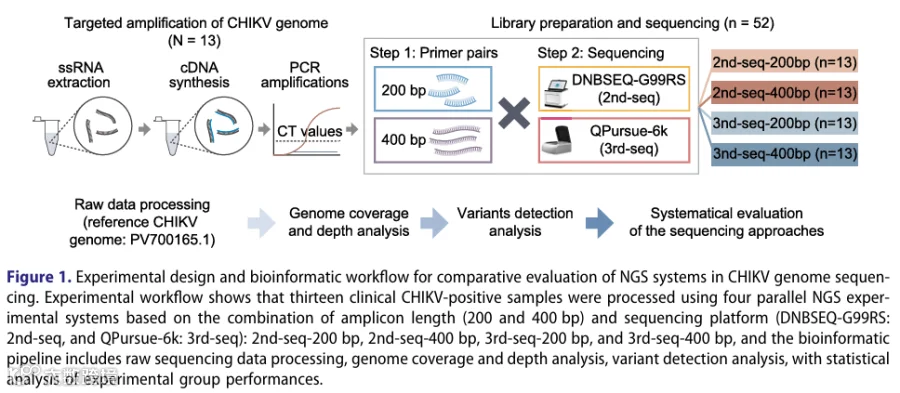
- 实验设计
采用13份Ct值跨度大的临床CHIKV阳性样本,平行构建四种NGS配置的文库(共52个文库):2nd-seq-200bp、2nd-seq-400bp、3rd-seq-200bp、3rd-seq-400bp。 - 数据生成
使用DNBSEQ-G99RS(2nd-seq)和QPursue-6k(3rd-seq)平台进行测序,获得12.6亿条通过质控的reads。 - 分析流程
以Réunion Island 80652-1-1-1为参考,用BWA/SAMtools比对,iVar进行变异识别和基因组组装。过滤条件包括最低深度≥10、质量≥20、等位基因频率≥3%,并进一步进行链偏差分析(Fisher精确检验,P<0.05)去除偏差变异。 - 评估指标
比较四种配置的测序深度、基因组覆盖率(单位点及滑动窗口)、变异数量、变异等位基因频率(VAF)分布、与Ct值的相关性、区域覆盖偏差、变异重叠情况,并进行系统发育分析验证一致性。 - Ct分层分析
根据Ct值对样本分层,分析不同深度阈值下基因组覆盖比例,并评估低覆盖区域对变异检测的影响。
研究亮点:
- 全面的平台比较
对四种NGS配置(2nd-seq-200bp, 2nd-seq-400bp, 3rd-seq-200bp, 3rd-seq-400bp)在同一批临床样本上进行平行评估。 - 低频变异检测能力
第三代测序(3rd-seq)检测到大量低频变异(VAF < 0.5),而第二代测序(2nd-seq)主要捕获高频共识变异。 - 低病毒载量样本表现
在Ct值>35的超低病毒载量样本中,第三代测序在变异检测方面优于第二代测序,覆盖更均匀。 - 覆盖均匀性优势
第三代测序提供了更均匀的基因组覆盖(99.9%),且对衣壳蛋白等困难区域的扩增偏差更具韧性。 - 平台特异性相关性
发现变异检测灵敏度与病毒载量(Ct值)存在平台依赖的相关性:2nd-seq呈负相关,3rd-seq呈正相关。
研究结果:
- 测序深度与产量
2nd-seq-400bp平均深度最高(152,430×),其次为2nd-seq-200bp(82,390×)、3rd-seq-400bp(62,729×)、3rd-seq-200bp(48,303×)。2nd-seq数据产量更高。 - 基因组覆盖
第三代测序平台实现更均匀覆盖(99.9% ± 0.3%),略优于第二代(99.4% ± 1.4%)。所有平台在衣壳蛋白编码区域均显示深度和覆盖降低,但第三代测序衰减更轻。 - 深度与Ct值相关性
2nd-seq深度与Ct值呈强负相关(200bp: r=-0.75; 400bp: r=-0.66),3rd-seq相关性较弱(200bp: r=-0.52; 400bp: r=-0.61)。 - 变异检测总体情况
经链偏差过滤后获得15,707个高置信度变异。3rd-seq-200bp检测到最多变异(1,553个唯一变异),3rd-seq-400bp为756个,而2nd-seq仅为416-417个。核心共有变异仅374个(20.66%)。 - VAF特征
2nd-seq变异VAF接近1.0(共识水平),而3rd-seq检测到大量低频变异(VAF<0.5),尤其在nsP1、nsP4、5'UTR、3'UTR和6K区域富集。 - Ct值与变异数量相关性
2nd-seq中变异数量与Ct值呈负相关(200bp: r=-0.69; 400bp: r=-0.76);3rd-seq中呈正相关(200bp: r=0.80; 400bp: r=0.76),提示低模板浓度下性能差异。 - 低病毒载量样本表现
在Ct>35样本中,3rd-seq从低覆盖区域(<500×)恢复44.54%-77.32%的变异,而2nd-seq仅0.85%-27.88%。 - 共识基因组分型
所有四种平台生成的共识基因组用于系统发育分析,结果完全一致,正确分配同一基因型,说明在基本分子流行病学应用中可靠性相同。 - 已知热点突变检测
所有平台均能可靠检测E1-A226V和E2-L210Q等已知流行病学相关突变。
研究总结:
结论:
-
第二代测序(尤其400 bp扩增子)在高通量、共识基因组生成方面最优,适合常规爆发监测。 -
第三代测序在低频变异发现、基因组覆盖均匀性、低病毒载量样本分析方面具有关键优势。 -
建议分层监测策略:Ct≤35样本两者皆可;Ct>35样本强烈推荐第三代测序进行深入分析。
讨论:
-
2nd-seq的平台深度极高,但区域特异性覆盖偏差(如衣壳区域)可能导致假阴性。 -
3rd-seq能揭示更丰富的宿主内病毒多样性,但需注意链偏差和低模板样本可能的技术伪影。 -
3rd-seq中变异数量与Ct值的正相关关系需要进一步研究,可能源于低覆盖区域驱动变异恢复,而非误差放大,但Ct>35样本的变异仍需谨慎解读。 -
两种平台在系统发育分型上的一致性表明,即使存在覆盖和变异检测差异,用于爆发溯源和基因型分类的共识基因组是可靠的。 -
研究为CHIKV爆发应对提供了实用工具,指出平台选择应取决于研究目标:群体水平监测选2nd-seq,深入表征选3rd-seq。
结果译文:
1.用于CHIKV基因组监测的NGS实验平台的系统评估
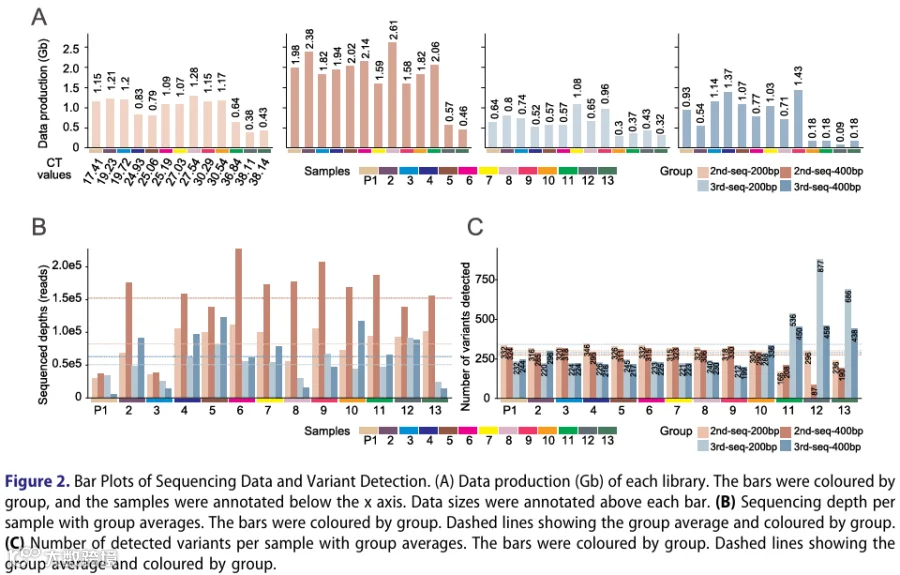
为确定常规CHIKV基因组监测的最佳测序条件,我们对四种下一代测序(NGS)配置进行了基准测试,这些配置将DNBSEQ-G99RS(二代测序)或QPursue6k(三代测序)化学方法与200 bp或400 bp扩增子引物组相结合(图1)。13份Ct值范围为17.41-38.14的临床样本被并行处理(图1)。共生成52个文库(每样本4个),获得12.6亿条通过质量控制(QC)的读段。我们发现DNBSEQ-G99(二代测序-400 bp)在数据产出效率上优于QPursue-6k(图2A,表1)。Ct值≥30.00的极低病毒载量样本,其数据产出量相对于其他样本有所减少(图2A)。经接头修剪、人源序列去除和CHIKV特异性比对后,保留的病毒读段用于下游深度和变异分析(图2B和C)。所有52个文库的蛋白质编码序列平均深度为87,108.24× ± 73,396.52×,其中二代测序-400 bp文库显示出最高的平均测序深度(152,430×),其次为二代测序-200 bp(82,390×)、三代测序-400 bp(62,729×)和三代测序-200 bp(48,303×)(图2B)。然而,变异检测在各组间呈现相对均匀的分布,平均变异数为302.06 ± 125.26,各组的平均变异数分别为276、289、302和342(图2C)。在这四种系统中,超低CHIKV载量样本(Ct值:P12: 38.11和P13: 38.14)尽管读段深度较低且高置信度变异数不一致,但仍产生了足够的读段深度用于变异检测(图2B和C)。总体而言,结果表明我们的实验系统即使在超低病毒载量样本中也能有效检测CHIKV。二代测序-400 bp系统产出更多数据和测序深度,其次是二代测序-200 bp、三代测序-400 bp和三代测序-200 bp。
2.三代测序用于CHIKV监测的均匀基因组覆盖度和对扩增偏倚的抗性
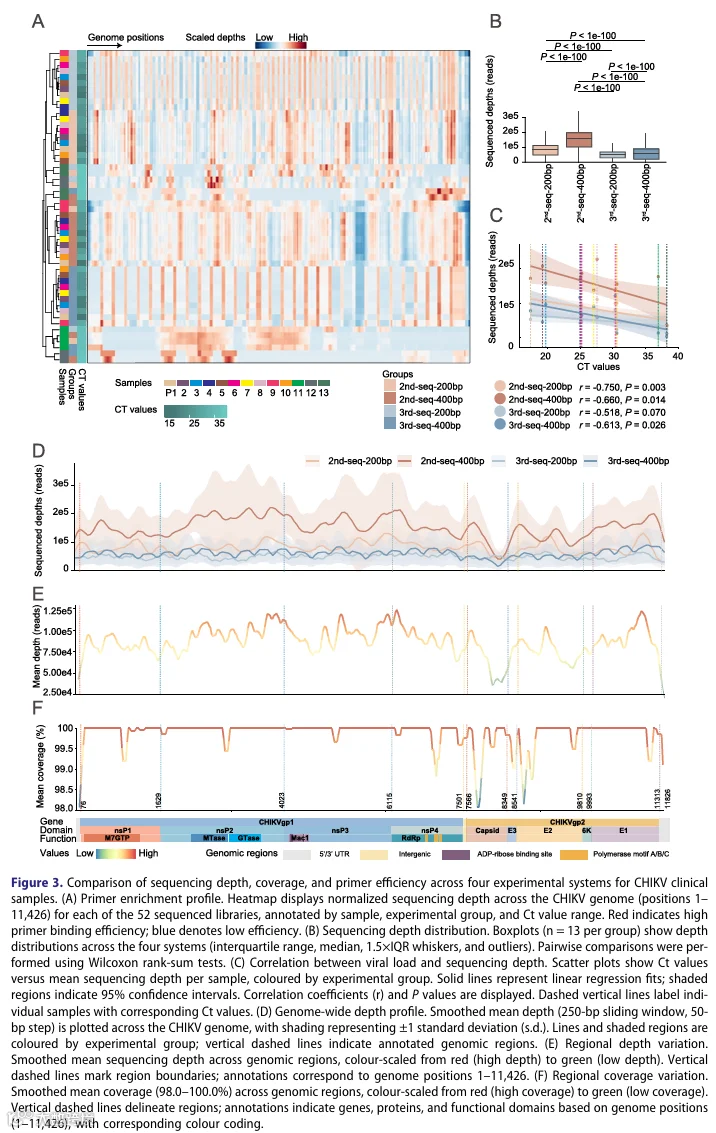
3.Ct分层覆盖结构对变异检测的影响
低病毒载量样本(Ct > 35)在增加的深度阈值下显示出急剧的覆盖度下降(图S4A)。三代测序从低覆盖区域(< 500×)中回收了44.54-77.32%的变异,而二代测序回收率低于20%,后者偏向于超深区域(> 10,000×)(图S4B-C)。相关性分析确认了在三代测序中,低质量样本的变异数与深度之间存在显著正相关(r = 0.30-0.34, p < 0.001,图S4D),表明低覆盖区域驱动了变异的回收——这一趋势在二代测序中未观察到。这些发现证明三代测序通过有效回收低覆盖区域的变异,在低病毒载量样本中捕获了更多的宿主内病毒多样性。
4.三代测序以更高的低频变异灵敏度解锁了CHIKV多样性的完整谱系
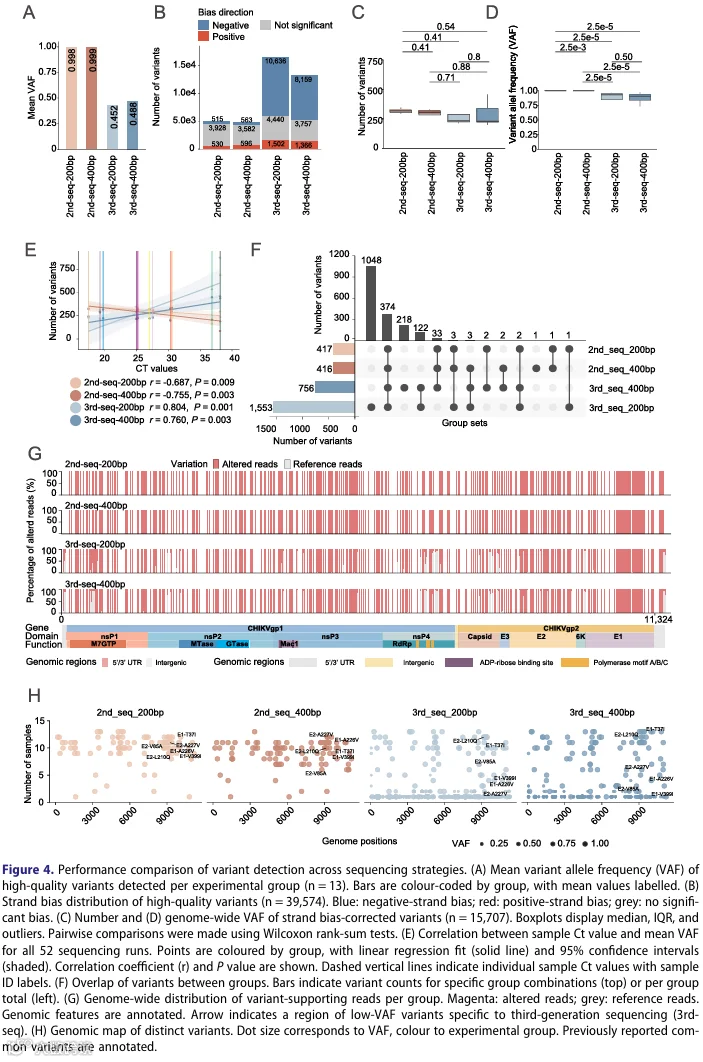
5.四种系统间检测独特变异的可靠性和一致性
6.所有四个测序平台的系统发育分型一致性
为评估测序平台或扩增子长度的选择是否影响分子流行病学应用,我们使用由四种方法的每一种为全部13份样本生成的共识基因组,构建了最大似然系统发育树。尽管在测序深度、覆盖均匀度和宿主内变异检测灵敏度方面存在差异,所有四个平台将每份样本置于相同的系统发育位置并分配了相同的基因型(补充图S3)。这种一致性证明,尽管平台在解析宿主内多样性方面的能力不同,但它们对于一级公共卫生应用(如疫情聚类、传播谱系追踪和基因型分类)是完全可靠的。
更多结果和补充图表:doi: 10.1080/22221751.2026.2660424



扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:https://cnsknowall.com
CNSknowall:24年最新问世的遥遥领先的颠覆性科研数据(0代码生信+统计学)分析平台,同时含有机制图模块(原创3000多素材和机制图模板)+AI一键生成高质量比国自然标书初稿+汉化版Pubmed融合Deepseek高效筛选目标文献同时一键提炼全文核心创新点+SCI文献例句/语料检索模块+全文翻译+文献求助+图片查重+期刊查询+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动发掘更多科研秘籍!