珊瑚白化是气候变暖引发的严重生态危机,但研究珊瑚全基因组(宿主+共生微生物)长期受困于宿主DNA污染(常>95%)和参考数据库不完整。本研究开发了holo-2bRAD技术——基于IIB型限制性内切酶BcgI的简化基因组测序方法。通过整合公共数据库中404,946个微生物基因组(395,422细菌+8,004古菌+1,505真菌)和宏基因组binning获得的56个珊瑚来源高质量MAG,构建了珊瑚全基因组参考数据库。技术验证显示:宿主与微生物2bRAD标签重叠率<0.3%,物种特异性达99.92%,技术重复基因分型一致性>99.9%,微生物检测Pearson相关系数>0.97。应用该技术分析不同白化程度的丛生盔形珊瑚,发现白化严重度与微生物α多样性呈负相关(Shannon指数,PERMANOVA p=0.003),有益菌Thermoanaerobacterium thermosaccharolyticum和Anaerosalibacter massiliensis在轻度白化中显著耗竭(log2FC分别为-25.2和-28.1),而三个未分类细菌随白化加重呈剂量依赖性富集。COG分析表明差异菌68%的基因涉及翻译功能。本研究为珊瑚全基因组高分辨率分析提供了新工具,有望推动基于微生物组的珊瑚保护策略。
今天给大家解读一篇4月发表在《Microorganisms》上的题目为“Holo-2bRAD: A Hologenomic Method for High-Resolution Analysis of Coral Microbiomes During Bleaching.”的文章。本文围绕“如何解决珊瑚宿主DNA污染导致微生物组分析困难”这一核心问题,提出并验证了holo-2bRAD技术。首先,通过整合公共数据库和自建binning结果,构建了包含404,946个微生物基因组和珊瑚宿主参考基因组的holo-DB,经BcgI酶模拟消化后保留99.92%物种的特异性标签。其次,对采集的Galaxea fascicularis样本(健康、轻度白化、严重白化)进行holo-2bRAD文库构建和测序,通过技术重复验证了方法的高重复性(物种检测100%一致,基因分型99.90%相似)。然后,利用该技术分析了不同白化程度珊瑚的微生物组:α多样性(Shannon指数)随白化加重显著下降(PERMANOVA p=0.003),有益菌(T. thermosaccharolyticum、A. massiliensis)在健康组富集,未解析细菌谱系剂量依赖性增加。同时,宿主SNP分析显示1619个SNP在组间共享,PCA显示健康组与白化组(B、C)聚类分离,但微生物变化与表型的相关性更强。结论认为holo-2bRAD为珊瑚全基因组研究提供了实用工具,支持基于微生物组的保护策略,并讨论了其局限性(缺乏空间分辨率、依赖数据库质量)和未来方向(与时FISH结合、持续扩充数据库)。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!包括数据集、绘图代码、图表复现、思路总结、参考文献……0代码!鼠标点点点即可轻松完成5-10分生信SCI全文复现!
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!



团队成员合影(位于上海陆家嘴中心,可随时预约参观)
题目:《Holo-2bRAD:一种全基因组方法,用于在珊瑚白化过程中对珊瑚微生物组进行高分辨率分析》Holo-2bRAD: A Hologenomic Method for High-Resolution Analysis of Coral Microbiomes During Bleaching
发表期刊:Microorganisms
影响因子:4.2
研究背景:
珊瑚礁被称为“海洋雨林”,支持约25%的海洋物种,但受到气候诱导白化的严重威胁。白化特征是珊瑚宿主与虫黄藻共生关系破裂,伴随微生物群落失调。传统方法中,扩增子测序存在高偏倚、低分辨率且无法同时检测细菌、古菌和真菌;宏基因组需要高质量DNA,不适用于宿主DNA污染严重(常>95%)的珊瑚样本。现有2bRAD-M技术已用于其他生物(如虾肠道菌群),但尚未应用于珊瑚全基因组研究。因此,亟需一种能够克服宿主污染、同时检测多域微生物并同步分析宿主遗传变异的高分辨率、低成本方法。
CNSknowall 平台 Pubmed+AI 快速提炼全文要点
研究思路:
- 数据库构建
从NCBI RefSeq和EnsemblFungi获取395,422细菌、8,004古菌、1,505真菌基因组,并结合从13个珊瑚宏基因组BioProject(370个样本)中通过MEGAHIT组装、MaxBin 2.0 binning、CheckM质量筛选(完整性>20%、污染<10%、基因组大小1-6 Mb)、GTDB-Tk分类注释获得的57个高质量MAGs。经BcgI酶模拟消化,保留含有唯一标签的基因组(404,628个),构建微生物特异性标签库。同时消化G. fascicularis宿主参考基因组(GCA_948470475.1),得到73,356个唯一宿主标签。将两者合并后进行二次冗余去除(移除0.90%珊瑚标签和2.15%微生物基因组),最终形成holo-DB。
- 样本处理与测序
采集亚龙湾不同白化程度的G. fascicularis样本,提取总DNA,按已发表protocol构建holo-2bRAD文库(包括BcgI酶切、接头连接、PCR扩增),在Illumina NovaSeq 6000上PE150测序。
- 数据分析
原始数据经质量过滤后,提取含BcgI识别位点的2bRAD标签;通过RADtyping进行宿主SNP基因分型(覆盖深度≥4×),构建NJ树和PCA;通过2bRAD-M流程比对holo-DB,进行微生物组成和相对定量分析;使用DESeq2鉴定差异物种,COG进行功能注释;计算α/β多样性并进行PERMANOVA检验。
- 性能验证
通过技术重复评估物种检测一致性、丰度相关性、基因分型一致性;通过比较海水与珊瑚样本验证数据库特异性。
研究亮点:
- 突破宿主DNA污染瓶颈
在技术重复中,宿主标签与微生物标签重叠极低(仅0.90%的珊瑚标签被移除),微生物映射率稳定(0.06%),实现了99.92%的物种级特异性,验证了在99%宿主污染样本中的可行性。
- 构建首个珊瑚专属全基因组数据库
整合395,422细菌、8,004古菌、1,505真菌参考基因组,以及从13个BioProject、370个样本中binning得到的57个高质量MAGs(56细菌+1古菌,平均完整性57.84%),显著扩展了对海洋共生微生物的覆盖。
- 极高的技术可重复性
两个技术重复在检测物种数量上100%一致,相对丰度Pearson r=0.9995,宿主基因分型一致性达99.90%,测序深度Pearson r>0.97,证实了文库构建和测序的稳定性。
- 揭示白化相关微生物标志物
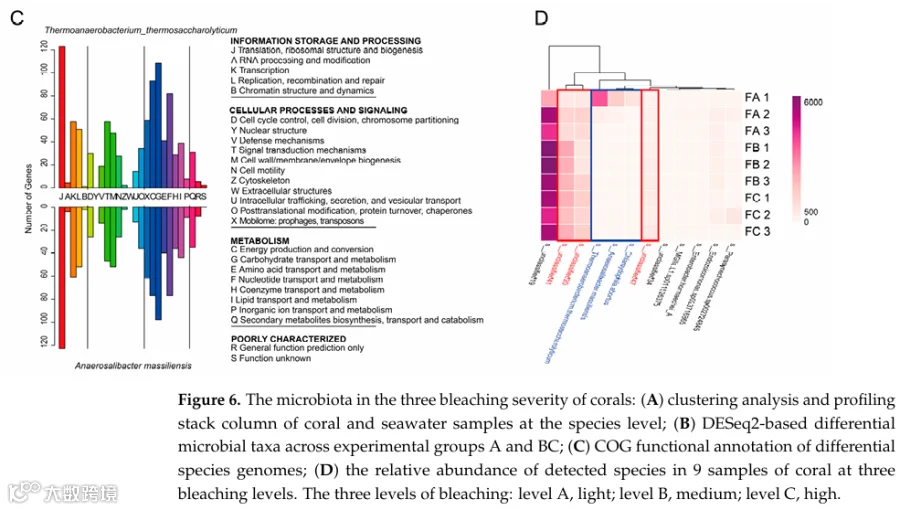
通过DESeq2鉴定出两个健康富集标志物(T. thermosaccharolyticum,log2FC=-25.2,p-adj=3.8×10⁻⁶;A. massiliensis,log2FC=-28.1,p-adj=2.1×10⁻⁸),其COG富集分析显示68%基因映射到翻译功能(Category J),提示蛋白质合成损伤可能是白化相关失调的功能后果。
研究结果:
- 数据库特征
404,628个微生物基因组被保留(99.92%初始基因组集),平均每个基因组产生1,264.955个标签;王国级标签特异度极高(门、科、属水平分别达99.70%、99.50%、99.10%);物种级51,834,062个限制性片段中99.92%为单拷贝标记;宿主标签73,356个,二次冗余移除后仅0.90%珊瑚标签被去除,对物种级影响可忽略(99.89%物种保留)。
- 技术重复性能
两个技术重复(Rep 1, Rep 2)获得可比原始读长(见表3),高质量读长比例>96%;宿主映射率19.99%-20.06%,微生物映射率稳定0.06%;检测物种数100%一致(图4A),相对丰度Pearson r=0.9995(图4B);宿主2bRAD位点覆盖率77.28%-77.29%,基因分型一致性99.90%,测序深度Pearson r>0.9706(图4C,D)。
- 宿主遗传变异
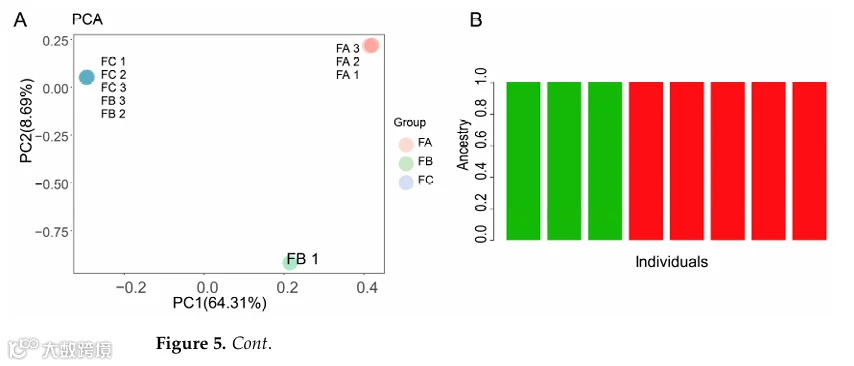
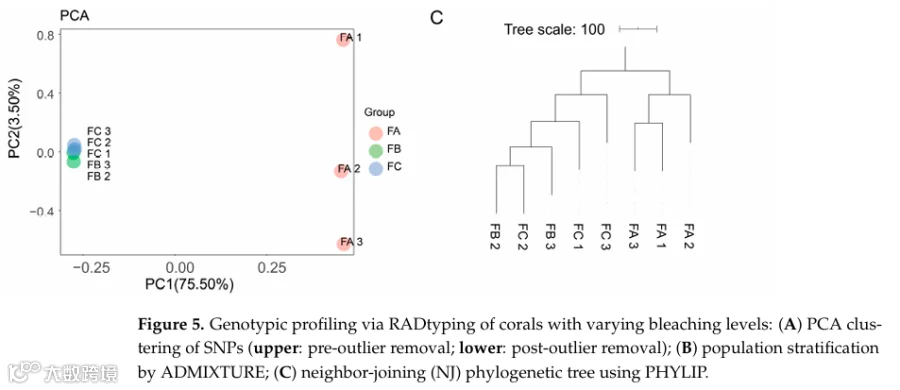
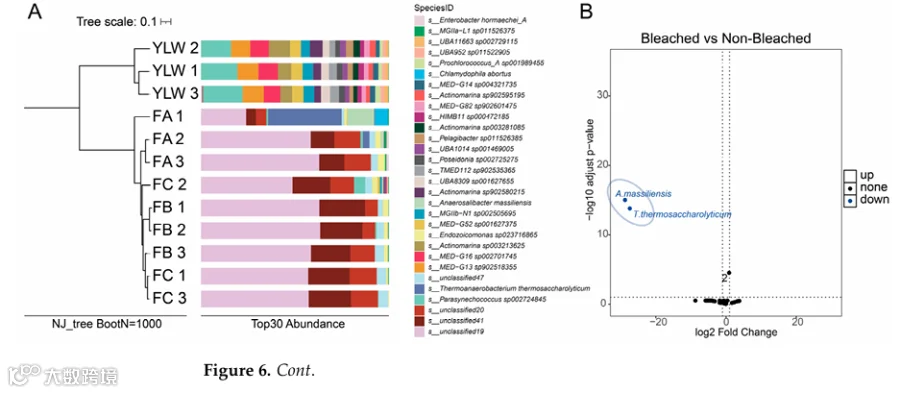
RADtyping检测到3915(Group A)、3585(Group B)、3440(Group C)个SNPs,其中1619个共有;PCA显示去除一个离群值后Group A独立聚类,Group B和C聚集(图5A);ADMIXTURE和NJ树进一步支持该分化(图5B,C)。
- 微生物群落动态
α多样性(Shannon指数)随白化加重显著下降(PERMANOVA p=0.003);优势科为Vallitaleaceae(42.38%-61.32%)和Pyrinomonadaceae(9.10%-22.51%);β多样性显示珊瑚微生物群与海水显著分离(Bray-Curtis=0.82,PERMANOVA R²=0.46,p=0.001)(图6A)。DESeq2鉴定两个健康富集标志物:T. thermosaccharolyticum(log2FC=-25.2, p-adj=3.8×10⁻⁶)和A. massiliensis(log2FC=-28.1, p-adj=2.1×10⁻⁸)(图6B);其COG功能注释中68%基因映射到翻译(Category J)(图6C)。三个未解析细菌谱系(s_unresolved19、20、41)在Group A中几乎检测不到,在Group B中少量出现,在Group C中显著富集(图6D)。
- 方法性能总结
数据库跨域干扰<0.3%,物种级标签保留99.92%唯一性;在0.06%微生物读长比例下仍可进行高分辨率分析;宿主SNP与微生物组成均检测良好。
研究总结:
结论:本文成功建立了holo-2bRAD技术,通过构建珊瑚专属全基因组数据库(Coral holo-DB),克服了高宿主DNA污染(~99%)的长期挑战,实现了对珊瑚全基因组(宿主+微生物)的单一文库、高分辨率分析。该方法在G. fascicularis白化梯度样本中验证了极佳的可重复性(技术重复100%物种一致)和特异性(99.92%),揭示了白化过程中微生物群落的有序重组:有益菌(如T. thermosaccharolyticum、A. massiliensis)显著减少,未解析细菌谱系剂量依赖性增加,且微生物变化与白化表型的相关性(α多样性下降p=0.003)强于宿主遗传变异(1619个共有SNP)。这为珊瑚白化机制研究提供了“微生物失调”新证据(COG富集分析提示翻译功能受损),并支持全基因组概念——认为珊瑚适应力由宿主与微生物共同决定。
讨论:
- 技术优势
与传统扩增子测序相比,holo-2bRAD无PCR偏倚、同时覆盖细菌/古菌/真菌/藻类;与宏基因组相比,成本低至1%、适用降解及高宿主污染样本。其构建的holo-DB覆盖范围超越现有以人类为中心的2bRAD-M数据库,且通过二次冗余去除实现了跨域最小干扰(<0.3%共享标签)。
- 生态学意义
健康珊瑚中富集的T. thermosaccharolyticum(嗜热解糖嗜热厌氧杆菌)和A. massiliensis可能是有益共生菌,其耗尽可能是白化的早期生物标志物。未解析谱系的剂量响应富集提示存在新的白化相关机会菌,需要后续分离培养和功能验证。宿主SNP在组间保守(1619个共有),而微生物组成却显著分化,表明白化表型差异更多由微生物失调驱动而非宿主遗传背景,这对传统的以宿主为中心的保育策略提出了挑战。
- 保护应用
方法稳健性(适用于降解样本、低生物量)使其适合大规模珊瑚礁健康普查;可基于holo-DB开发环境DNA(eDNA)监测,通过海水样本非侵入性追踪微生物信号;为益生菌干预提供高分辨率数据以识别候选菌株并监测效果;结合多组学数据(SNP+COG)可揭示宿主-微生物互作分子机制。
- 局限性
作为批量DNA测序方法,缺乏空间分辨率(无法定位微生物的具体解剖位置如黏液层、胃皮组织等);数据库完整性依赖于参考基因组质量,新谱系(如未解析的3条菌株)可能无法精准鉴定。未来需结合FISH等空间技术,并持续扩充数据库(如加入更多珊瑚共生藻Symbiodiniaceae基因组)。
- 总结
holo-2bRAD为珊瑚全基因组研究提供了“实用、可扩展、高分辨率”的平台,有望将珊瑚礁保护从被动恢复转向主动、数据驱动的韧性管理,助力理解并缓解气候变化对珊瑚礁的影响。
结果译文:
holo-2bRAD方法的实际应用——能够使用单个测序文库对珊瑚全基因组进行纵向分析——依赖于三个基本条件。第一,珊瑚样本的采集,包括用于比较分析的健康和白化标本;第二,同时提取包含宿主及其共生微生物遗传信息的复合基因组物质,这是整合全基因组分析的基本要求;最后,开发包含宿主特异性和微生物分类单元特异性限制性标签的定制参考数据库,使得尽管在文库中共同扩增,仍能对测序读段进行精确的生物信息学区分。整个工作流程的概述如图1所示。在计算分析工作流程中,第一步是构建珊瑚全基因组数据库,该数据库包含微生物分类单元特异性和宿主特异性的2bRAD标签。构建该数据库的详细步骤见第2节。随后,原始测序读段进行预处理,之后将高质量读段比对到全基因组数据库,以检索用于宿主基因分型和微生物群分析的可靠读段。最终,宿主来源的测序2bRAD标签通过RADtyping程序进行基因分型,以获取宿主的全基因组基因型。相反,微生物来源的标签用于使用2bRAD-M流程进行微生物组成分析和相对定量,这与我们之前在扇贝和虾类研究中应用的方法一致。
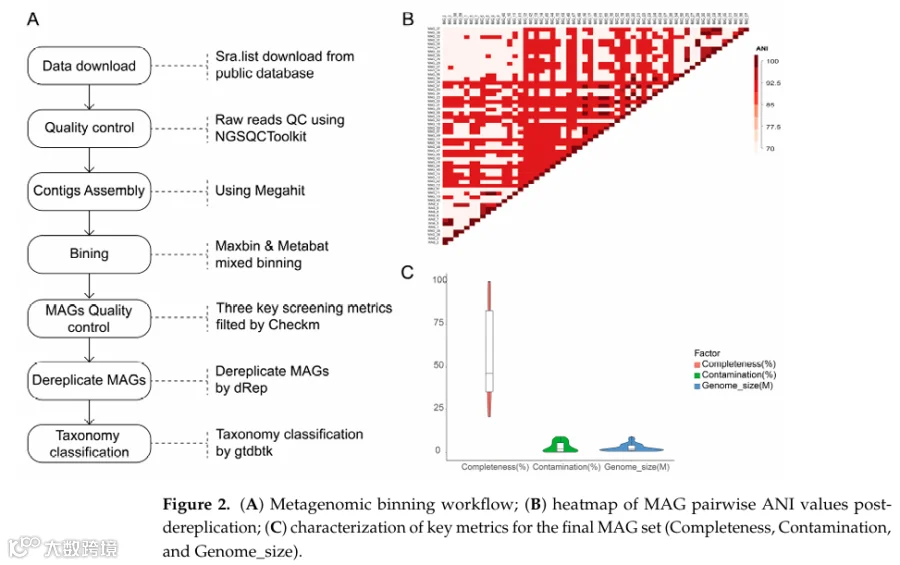
为了进一步提高定制holo-2bRAD参考数据库的全面性、代表性和分类覆盖度,我们整合了公开的珊瑚相关宏基因组数据,并进行了系统的宏基因组组装和分箱分析。共有13个独立BioProjects(保存在NCBI SRA数据库中,涵盖370个个体生物样本)被纳入这一整合分箱分析(图2)。使用MEGAHIT流程进行宏基因组序列组装,随后使用默认参数的MaxBin 2.0分箱流程从这些组装的珊瑚宏基因组数据集中恢复MAG。为了消除冗余或高度相似的基因组bin,并确保在物种水平上的非冗余代表性,我们计算了所有恢复的MAG之间的两两平均核苷酸一致性值,并在>97% ANI的阈值下进行去冗余,这是界定原核生物物种的广泛接受标准。宏基因组组装、分箱、去冗余和质量过滤的详细工作流程总结于图2A。
使用CheckM进行MAG质量评估,以估计基因组完整性和潜在污染。对于后续分析和数据库构建,仅保留满足以下条件的高质量MAG:完整性>20%,污染<10%,基因组大小在1至6 Mb之间。应用此严格的质量过滤程序后,我们最终获得了57个适合数据库整合的高质量MAG。这些合格的MAG平均完整性为57.84±26.73%,平均污染水平为3.14±2.91%,平均基因组大小为2.55±2.07 Mb(表1)。
为了确定这些分箱微生物基因组的分类学身份和系统发育分布,我们使用基因组分类数据库工具包v2.4.0进行了标准化的分类学注释。总体而言,宏基因组分箱分析产生了57个高质量微生物基因组bin,包括56个细菌基因组和1个古菌基因组。这些MAG代表了广泛的分类多样性,跨越9个不同的门、15个目和24个属,其中5个MAG被可靠地分类到物种水平(图2C;表2)。这些分类多样化的MAG的恢复显著扩展了我们holo-2bRAD参考数据库的深度和广度,从而提高了下游微生物分析和分类分配的准确性。
为了确保准确和稳健的下游分析,必须首先按照我们先前建立的方案构建物种特异性的全基因组数据库。该数据库包含高置信度的限制性内切酶消化标签,能够同时进行微生物群落的高分辨率分类学分析和珊瑚宿主的基因分型。
为此,共检索了404,946个微生物基因组用于数据库构建。该基因组集合包括从我们的宏基因组测序数据中恢复的57个非冗余MAG,以及从NCBI RefSeq和EnsemblFungi数据库获取的395,422个细菌、8,004个古菌和1,505个真菌完整基因组。然后使用IIB型限制性内切酶BcgI对所有收集的基因组进行计算机模拟限制性消化。
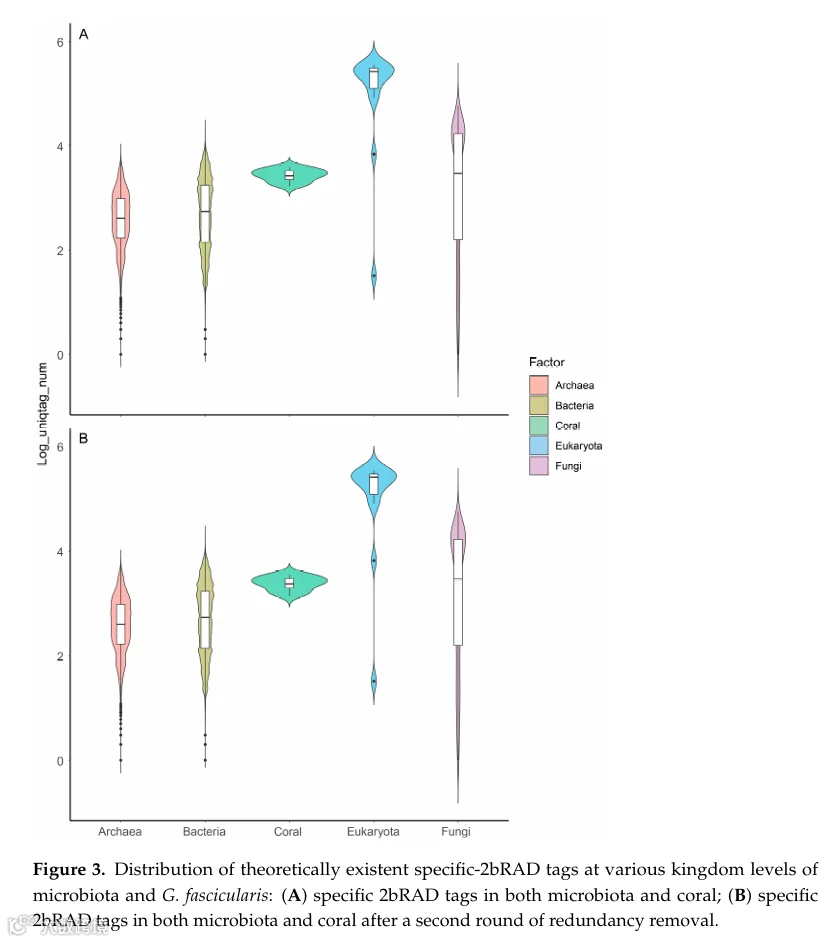
消化步骤后,共保留了404,628个基因组(占整个初始基因组集的99.92%);其余基因组因缺乏BcgI识别位点或生成的标签不唯一而无法可靠区分被排除。这些保留的基因组随后被整合到基础holo-2bRAD数据库中,每个基因组平均生成1,264.955个标签。从这些生成的标签中,选择目标holo-2bRAD标签——定义为每个基因组中单拷贝存在的微生物分类单元特异性DNA标记——予以保留。总体而言,在不同的分类等级上,一个明显的趋势是:分类等级越高,可保留的可用标签数量越多(图3A)。在界水平上,细菌、真菌和古菌之间的holo-2bRAD标签共享极少;因此,几乎所有的holo-2bRAD标签都表现出界特异性。具体而言,门特异性、科特异性和属特异性的holo-2bRAD标签分别占单个微生物基因组产生的所有理论holo-2bRAD标签的99.70%、99.50%和99.10%。
我们进一步评估了86,902个物种水平微生物类群中holo-2bRAD标签的独特性。在BcgI消化产生的51,834,062个总限制性片段中,约99.92%作为其各自微生物基因组中的单拷贝标记存在。同时也在G. fascicularis珊瑚宿主基因组上进行了计算机模拟消化。获得了111,748个初始非冗余宿主标签,经过严格的第二轮去冗余后,精炼为73,356个高置信度独特宿主标签。
对于最终的全基因组数据库构建,将微生物分类单元特异性标签和宿主独特标签合并,并进行第二轮去冗余。通过这一步,仅去除了658个珊瑚来源的标签(占所有非冗余标签的0.90%)和约8,689个微生物基因组(占微生物基因组总数的2.15%)(图3B)。这些微生物基因组的去除对从属到门的分类水平影响可忽略不计(例如,影响率为0.01%至0.1%)。值得注意的是,在数据库构建过程中保留了86,902个物种(占总物种的99.89%);这一观察表明,上述微生物基因组的去除在物种水平上未产生显著影响。
综合这些发现,珊瑚宿主与其相关微生物之间的标签重叠极少。这种极少的重叠对珊瑚的基因分型或其相关微生物组的分析几乎没有影响。反过来,这一结果证明了使用单个holo-2bRAD文库进行全基因组分析的可行性。
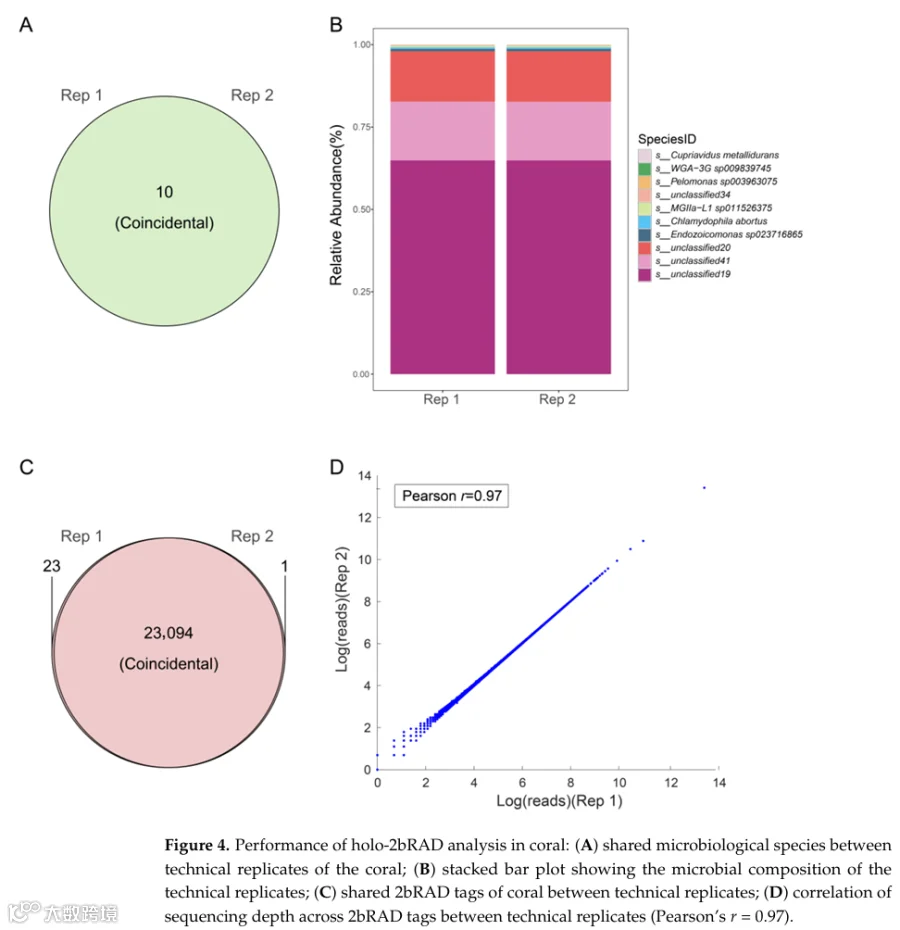
为了验证holo-2bRAD在珊瑚全基因组分析中的可靠性和稳定性,我们构建了一组技术重复文库(Rep 1和Rep 2)。对这些文库的测序产生了相同物种可比的原始读段数量(表3)。经过严格的质量控制过滤后,超过96%的测序读段被保留为用于下游分析的高质量读段。携带BcgI识别位点的高质量读段被比对到珊瑚全基因组数据集。在两个技术重复中,比对到珊瑚宿主基因组的比率在19.99%至20.06%之间,而比对到相关微生物群落的比率稳定在0.06%,表明文库构建和测序性能高度一致。
为了评估我们的方法在表征珊瑚相关微生物群落方面的一致性,我们对可重复性进行了评估。在两个技术重复之间,检测到的物种数量高度相似(100%)(图4A)。在相对丰度分析中,两个重复之间检测到的物种丰度没有显著差异(图4B),相关性分析(Pearson's r = 0.9995)表明了这一点。这种高可重复性证明了宏基因组分析的稳定性能。
除了微生物分析,我们还系统评估了holo-2bRAD用于珊瑚宿主基因分型的性能,重点关注四个关键参数:全基因组标签覆盖率、基因型检出率、技术重复之间的基因型一致性以及基因型准确性。对检测到的2bRAD位点全基因组覆盖率的检查显示,所有样本的覆盖率高度均匀,两个技术重复的值分别为77.28%和77.29。基因型检出(显示99.90%的相似性)以及每个2bRAD标签的测序深度在重复之间都表现出卓越的一致性。这种高重复间一致性还得到Pearson相关系数大于0.9706的支持(图4C、D),共同验证了holo-2bRAD用于全基因组宿主基因分型的稳健稳定性和精确性。
为了进一步验证holo-2bRAD在珊瑚全基因组研究中的实际应用性,我们分析了从未白化、轻度白化和严重白化的G. fascicularis珊瑚群体生成的holo-2bRAD文库,以及在同一次白化事件期间收集的海水参考样本(表3)。比对到定制全基因组数据库的结果显示,不同白化严重程度的珊瑚样本之间微生物比对率存在明显差异(表4)。RADtyping在A、B和C组中分别检测到3,915、3,585和3,440个SNP,其中1,619个在三组间共享。SNP数据的PCA显示B组中有一个离群值(归因于个体因素);排除后,A组形成一个独特簇,而B组和C组则聚集在一起(图5)。
分析了不同白化水平的珊瑚相关微生物群以及海水参考样本。白化严重程度较低的珊瑚具有显著更高的微生物α多样性(Shannon指数,PERMANOVA,p=0.003)。Vallitaleaceae(分别为42.38±17.16%、61.32±2.48%、53.63±5.97%)和Pyrinomonadaceae(分别为9.10±3.66%、22.51±1.53%、21.26±1.71%)是优势菌科。海水微生物群的β多样性与珊瑚相关群落明显不同(Bray-Curtis不相似度=0.82,PERMANOVA,R²=0.46,p=0.001;图6A)。DESeq2鉴定出两个生物标志物:Thermoanaerobacterium thermosaccharolyticum(A组富集,log2FC=-25.2,p-adj=3.8×10⁻⁶)和Anaerosalibacter massiliensis(A组富集,log2FC=-28.1,p-adj=2.1×10⁻⁸;图6B)。COG富集分析显示,它们68%的基因映射到翻译类别(J类;图6C)。
三个未解析的细菌谱系(s_unresolved19、s_unresolved20和s_unresolved41)表现出与白化严重程度增加相关的剂量反应性富集(图6D)。
更多结果和补充图表:doi:10.3390/microorganisms14040840
长按二维码关注我们,用最短的时间和最高的效率学习更多生信思路!
扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:https://cnsknowall.com
CNSknowall:24年最新问世的遥遥领先的颠覆性科研数据(0代码生信+统计学)分析平台,同时含有机制图模块(原创3000多素材和机制图模板)+AI一键生成高质量比国自然标书初稿+汉化版Pubmed融合Deepseek高效筛选目标文献同时一键提炼全文核心创新点+SCI文献例句/语料检索模块+全文翻译+文献求助+图片查重+期刊查询+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动发掘更多科研秘籍!