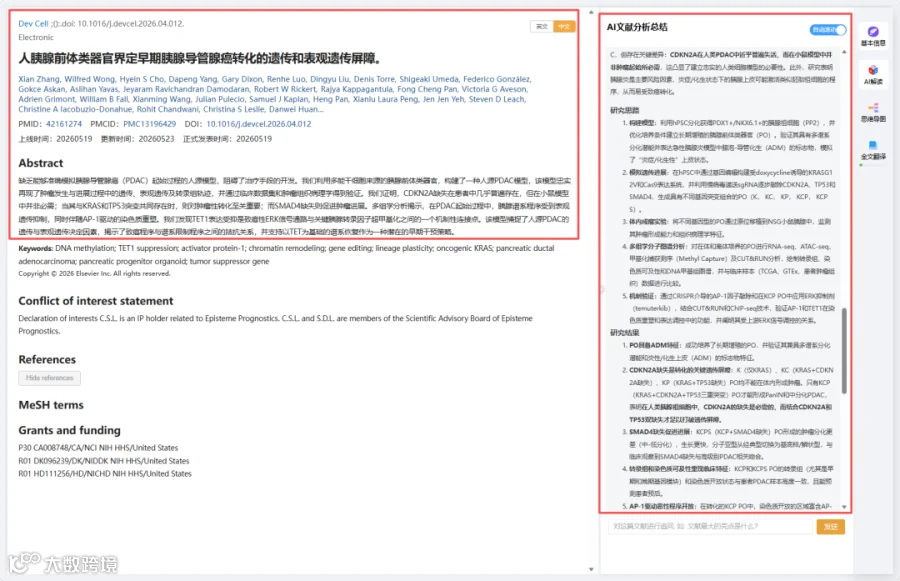
胰腺导管腺癌(PDAC)致死率高,缺乏能忠实再现人肿瘤发生早期事件的模型。本研究利用人 pluripotent stem cell(hPSC)来源的胰腺祖细胞类器官(PO),结合CRISPR基因编辑(依次引入KRASG12V、CDKN2A敲除、TP53敲除及SMAD4敲除),建立了一个人源PDAC转化模型。通过RNA-seq、ATAC-seq、CUT&RUN及EPIC甲基捕获测序等多组学生信分析,并与TCGA-PAAD、GTEx及单细胞公共数据库比较,发现:KRAS+CDKN2A+TP53三重突变足以驱动PO形成PanIN和中分化PDAC,而CDKN2A或TP53单独缺失均不足以转化。机制上,致癌性ERK信号激活AP-1(FOSL2/FOS/JUNB)驱动恶性表观重编程,同时抑制TET1导致胰腺谱系转录因子(ONECUT1、HNF1B)高甲基化沉默。本研究揭示了人PDAC转化的遗传和表观遗传双重屏障。
今天给大家解读一篇5月发表在《Developmental Cell》上的题目为“Human pancreatic progenitor organoids define genetic and epigenetic barriers to early PDAC transformation.”的文章。本研究通过从人多能干细胞分化并培养胰腺前体类器官,利用CRISPR/Cas9逐步引入临床相关的PDAC驱动基因突变(KRASG12V, CDKN2A缺失, TP53缺失, SMAD4缺失)。研究发现,只有同时具有KRAS、CDKN2A和TP53三重突变的类器官(KCP PO)才能在异种移植后形成胰腺上皮内瘤变(PanIN)和中分化PDAC。额外的SMAD4缺失(KCPS PO)则导致快速进展为低分化、基底样亚型PDAC。多组学分析(RNA-seq、ATAC-seq、甲基化捕获、CUT&RUN)揭示了转化过程中染色质可及性的双相变化:AP-1转录因子(如FOSL1/2、FOS、JUNB)驱动的恶性基因位点开放,以及TET1降低导致的胰腺谱系转录因子(如ONECUT1、PDX1、HNF1B)位点的甲基化和关闭。ERK信号通路是调控这一平衡的关键上游信号,抑制ERK可逆转部分恶性特征并恢复谱系特征。研究还发现这些分子特征与临床患者样本的病理分级和分子亚型高度一致。(请持续关注我们,每天为您解读最新见刊的文献!)想薅生信资料羊毛?直接在对话框回复 “资料”,免费领取干货大礼包!包括数据集、绘图代码、图表复现、思路总结、参考文献……0代码!鼠标点点点即可轻松完成5-10分生信SCI全文复现!
不想做实验,没数据,还想要快速发表文章,没问题的!公共数据库就是我们的数据宝藏!没思路不用担心,作为专业的生信团队,我们很乐意为你们效劳,提供研究路线设计和数据挖掘分析,扫码联系我们吧!



团队成员合影(位于上海陆家嘴中心,可随时预约参观)
题目:《人胰腺前体类器官界定早期胰腺导管腺癌转化的遗传和表观遗传屏障》Human pancreatic progenitor organoids define genetic and epigenetic barriers to early PDAC transformation
发表期刊:Developmental Cell
影响因子:8.7
研究背景:
胰腺导管腺癌诊断时多为晚期,预后极差,其早期癌变机制尚不明确。临床样本分析显示,KRAS、CDKN2A、TP53和SMAD4的突变在PanIN阶段就已出现,提示这些遗传事件是转化的驱动力。虽然基因工程小鼠模型(GEMM)已用于研究PDAC,但存在关键差异:CDKN2A在人类PDAC中近乎普遍失活,而在小鼠模型中并非肿瘤起始所必需,这凸显了建立忠实的人类细胞模型的必要性。此外,研究表明胰腺炎是主要风险因素,炎症/化生状态下的胰腺上皮可能激活类似胚胎祖细胞的程序,从而易受致癌转化。
CNSknowall 平台 Pubmed+AI 快速提炼全文要点
研究思路:
- 构建模型
利用hPSC分化获得PDX1+/NKX6.1+的胰腺祖细胞(PP2),并优化培养条件建立长期增殖的胰腺前体类器官(PO)。验证其具有多谱系分化潜能并表达急性胰腺炎模型中腺泡-导管化生(ADM)的标志物,模拟了“炎症/化生性”上皮状态。
- 模拟遗传进展
在hPSC中通过基因编辑构建受doxycycline诱导的KRASG12V和Cas9表达系统,并利用慢病毒递送sgRNA逐步敲除CDKN2A、TP53和SMAD4,生成具有不同基因突变组合的PO(K, KC, KP, KCP, KCPS)。
- 体内成瘤实验
将不同基因型的PO通过原位移植到NSG小鼠胰腺中,监测其肿瘤形成能力和组织病理学特征。
- 多组学分子图谱分析
对在体和离体培养的PO进行RNA-seq、ATAC-seq、甲基化捕获测序(Methyl Capture)及CUT&RUN分析,绘制转录组、染色质可及性和DNA甲基组图谱,并与临床样本(TCGA、GTEx、患者肿瘤组织)数据进行比较。
- 机制验证
通过CRISPR介导的AP-1因子敲除和在KCP PO中应用ERK抑制剂(temuterkib),结合CUT&RUN和ChIP-seq技术,验证AP-1和TET1在染色质重塑和表达调控中的功能,并阐明其受上游ERK信号调控的关系。
研究亮点:
- 建立了忠实再现人类PDAC起始过程的类器官模型
利用hPSC来源的胰腺前体类器官,通过组合引入驱动基因突变,成功在体内外再现了从PanIN到不同分化程度PDAC的转化,其分子特征与临床患者数据高度一致。
- 揭示了CDKN2A在人类PDAC转化中的必要性
与小鼠模型不同,本研究表明仅KRAS激活并联合单个TSG(CDKN2A或TP53)缺失不足以导致肿瘤。CDKN2A的缺失是与KRAS和TP53突变共同驱动人类PDAC起始的必需条件,填补了小鼠模型与临床观察之间的重要鸿沟。
- 阐明了早期PDAC转化的双向表观遗传编程机制
研究发现了同时发生的两个对立过程:AP-1转录因子驱动的、促进恶性特征的表观遗传激活,以及TET1介导的、维持正常胰腺谱系身份的表观遗传沉默。这揭示了“致癌程序激活”与“谱系限制程序丧失”之间的拮抗作用。
- 提出基于TET的谱系恢复作为潜在早期干预策略
鉴于TET1的丧失是谱系抑制的关键环节,研究结果支持通过恢复TET活性或重新激活胰腺谱系表达作为针对高危人群的早期干预新策略。
研究结果:
- PO具备ADM特征
成功培养了长期增殖的PO,并验证其兼具多谱系分化潜能和炎性/化生上皮(ADM)的标志物特征。
- CDKN2A缺失是转化的关键遗传屏障
K(仅KRAS)、KC(KRAS+CDKN2A缺失)、KP(KRAS+TP53缺失)PO均不能在体内形成肿瘤。只有KCP(KRAS+CDKN2A+TP53三重突变)PO才能形成PanIN和中分化PDAC,表明在人类胰腺祖细胞中,CDKN2A的缺失是必需的,而结合CDKN2A和TP53双缺失才足以打破遗传屏障。
- SMAD4缺失促进进展
KCPS(KCP+SMAD4缺失)PO形成的肿瘤分化更差(中-低分化),生长更快,分子亚型从经典型切换为基底样/鳞状型,与临床观察到SMAD4缺失与高级别PDAC相关吻合。
- 转录组和染色质可及性重现临床特征
KCP和KCPS PO的转录组(尤其是早期和晚期基因模块)和染色质开放状态与患者PDAC样本高度一致,且能预测患者预后。
- AP-1驱动恶性程序开放
在转化的KCP PO中,染色质开放的区域富含AP-1(特别是FOSL1/2、FOS、JUNB)的基序。敲低这些AP-1因子会抑制KCP PO的增殖和恶性标志物表达。CUT&RUN证实这些AP-1因子直接结合并激活恶性相关基因的调控区域。
- TET1丧失介导谱系程序关闭
在转化的PO中,TET1(而非TET2/3)表达下调,导致全局5hmC水平降低和胰腺谱系转录因子(如ONECUT1, PDX1)基因位点发生超甲基化和染色质关闭。该现象在患者样本的单细胞分析中也得到验证。
- ERK信号整合两个过程
ERK抑制剂处理KCP PO,同时抑制了AP-1介导的染色质开放(恶性程序)和恢复了TET1结合位点的染色质可及性(谱系程序),证实ERK是上游关键调控枢纽。
研究总结:
结论:
-
该研究成功建立了一个基于人胰腺前体类器官的PDAC模型,该模型忠实再现了人类PDAC从早期转化到进展的遗传、表观遗传和转录组轨迹,且与临床数据高度契合。
- 遗传层面
明确了CDKN2A在与KRAS和TP53协同作用下,是驱动人类PDAC起始的独特且关键的遗传屏障(不同于小鼠模型)。SMAD4缺失则推动肿瘤向高级别、高侵袭性方向进展。
- 表观遗传层面
揭示了早期PDAC转化涉及两个ERK信号调控下的对立过程:a) 通过激活AP-1转录程序进行“获得性”的致癌重编程;b) 通过抑制TET1导致谱系特异性转录因子超甲基化,从而发生“丧失性”的谱系分化屏障崩溃。
- 治疗意义
该模型为解析PDAC的早期事件提供了强大平台,并首次提出通过恢复TET1功能或胰腺谱系表达的谱系修复疗法,可能成为针对高危人群的早期干预新策略。
讨论:
- 模型优势与局限性
该模型通过利用胚胎胰腺祖细胞模拟了胰腺炎后的炎症/化生上皮状态,成功复现了人类PDAC的遗传要求,这可能是其与以往使用成熟导管/腺泡细胞模型结果不一致的关键。一个显著局限是,体外培养缺乏天然的肿瘤微环境,这可能导致了无法模拟低级别PanIN的缓慢进展,以及K/KC/KP基因型PO的致瘤失败。
- 与小鼠模型的差异
本研究结果明确指出了人和小鼠在PDAC转化中的根本差异:人类细胞对致癌转化更具抵抗力,需要更完全(如CDKN2A+TP53双缺失)的遗传打击才能打破屏障。这高度强调了使用人类模型研究人类疾病的必要性。
- 肿瘤可塑性机制
研究将PDAC中观察到的肿瘤可塑性解构为“恶性转变”(获取AP-1驱动的癌特征)和“身份丧失”(TET1依赖的谱系抑制)两个紧密耦合的表观遗传过程。这为理解肿瘤进展和耐药中的细胞身份转变提供了新的分子框架。
- 早期干预的潜力
与靶向已经形成的肿瘤中特定基因突变不同,本研究提出的“谱系恢复”策略旨在通过治疗性干预,逆转早期肿瘤细胞中的表观遗传变化,恢复其正常分化状态。这为高危人群的预防和早期治疗开辟了新的可能性。
结果译文:
1.胰腺祖细胞类器官维持谱系潜能并重现ADM的标志性特征
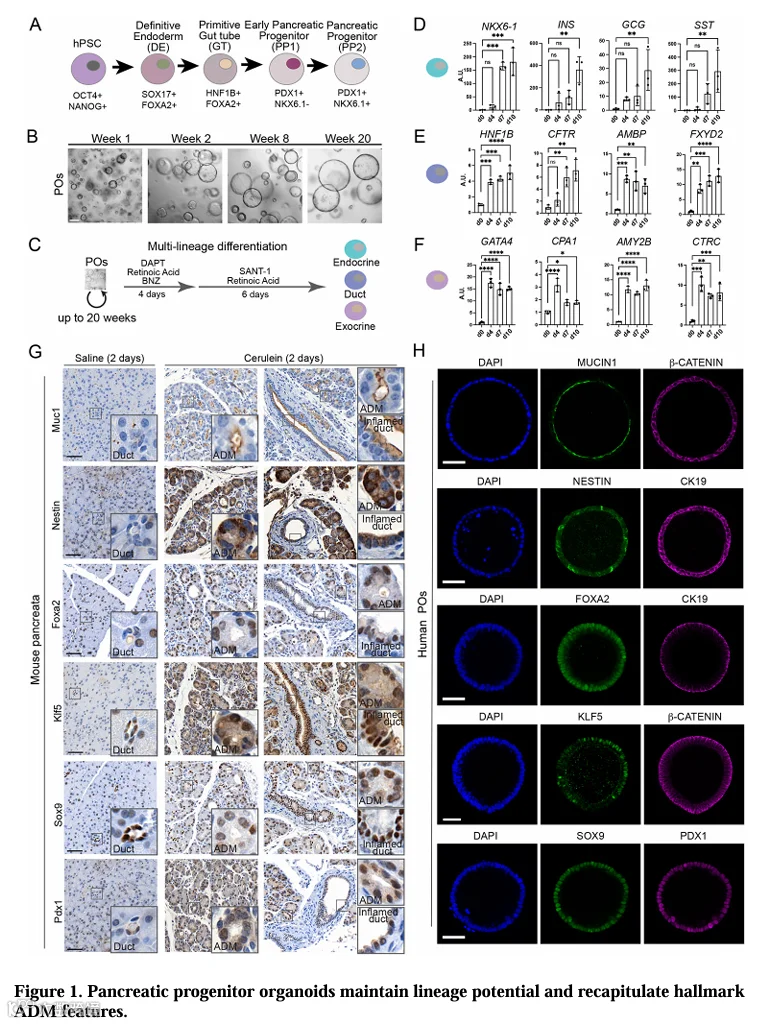
我们从hPSC通过定向分化平台生成了PDX1+/NKX6.1+的PP2细胞(图1A,S1A)。为了实现基因编辑和分子表征,我们优化了扩增条件[22-25],并建立了一种3D胰腺祖细胞类器官(PO)培养体系,该体系能支持祖细胞长期增殖超过20周(图1B)。在扩增过程中,PO维持了早期胰腺祖细胞(PP1)标志物PDX1和SOX9的表达,但失去了在PP1向PP2转变过程中获得的NKX6.1表达,这与既往报道一致(图S1B,C)[25]。重要的是,扩增中的类器官保留了多能性,通过多谱系分化得以证明(图1C),该分化重新激活了NKX6.1的表达,并诱导了胰腺内分泌(INS、GCG、SST)、导管(HNF1B、CFTR、FYD2、AMBP)和腺泡(GATA4、CPA1、AMY2B、CTRC)标志物的表达,经RT-qPCR和免疫荧光验证(图1D-F,S1D,E)。
为了评估这些PO是否能重现炎症状态下的胰腺上皮,我们将其与雨蛙素诱导的急性胰腺炎小鼠模型进行了比较。雨蛙素处理导致Muc1、Nestin、Foxa2、Klf5在腺泡-导管化生(ADM)和炎症导管中相对于正常导管上调,而免疫荧光证实了这些已建立的ADM标志物在hPSC来源的PO中的表达(图1G,H)。Sox9和Pdx1(强标记小鼠ADM和炎症导管)在PO中也大量存在(图1G,H)。这些结果表明,PO保留了多能祖细胞程序,并重现了ADM和炎症胰腺上皮的标志性特征,为探究PDAC转化的最早步骤提供了人细胞背景。
2.致癌性KRAS联合单一CDKN2A或TP53缺失不足以启动PDAC
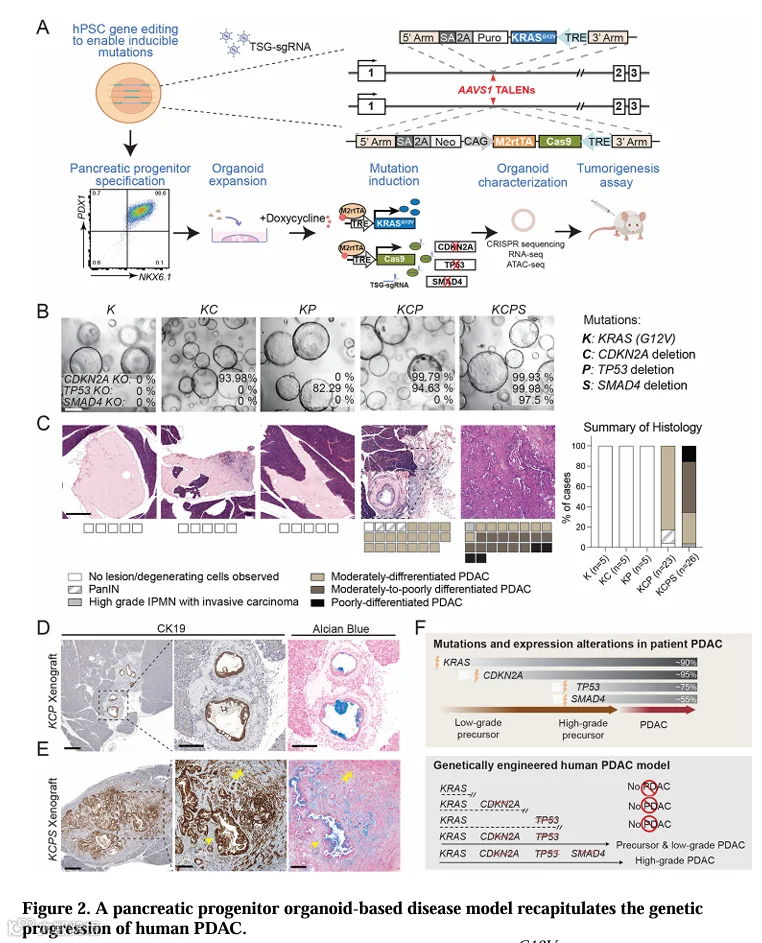
为了检验CDKN2A和TP53突变联合KRAS是否足以像小鼠模型那样驱动PDAC,我们设计了携带强力霉素诱导的GFP标记KRASG12V和Cas9元件的hPSC,以实现可控的癌基因激活和TSG失活,同时构建了表达GFP和Cas9的匹配细胞系(图2A,S2A,B)。通过慢病毒向KRASG12V/Cas9诱导型hPSC递送靶向CDKN2A或TP53的sgRNA,建立了一个可诱导激活致癌性KRAS并同时删除TSG的系统(图2A)。
这些经过基因编辑的hPSC被分化为PP2阶段,效率与野生型细胞相当(>90% PDX1+/NKX6.1+),然后转入PO培养,并用强力霉素处理以激活致癌性KRAS表达并驱动TSG缺失,从而产生K、KC和KPPO(K:KRASG12V,C:CDKN2A缺失,P:TP53缺失)(图2A)。CRISPR测序显示,在诱导后7周,KC和KPPO中分别以高效率实现了CDKN2A和TP53位点的敲除(定义为移码或>21 bp缺失),并通过Western blot证实(图2B,S2E)。值得注意的是,所有表达KRASG12V的PO均以极化囊泡的形式增殖,未出现先前报道的“管腔填充”表型[26,27],这可能反映了起始细胞身份、培养条件或癌基因剂量的差异。
为了评估致瘤能力,将基因型验证后的K、KC和KPPO(培养约20周)原位移植到饮用水中含强力霉素的NSG小鼠体内,并监测肿瘤形成长达6个月(图2A)。尽管致癌性KRAS持续激活,但在注射K、KC和KPPO的动物中未检测到上皮病变。相反,偶尔观察到瘢痕反应和退变细胞营养不良性钙化,表明移植物未成功定植(每种基因型n=5,图2C,S2F,G)。因此,与小鼠模型相反[15,28],致癌性KRAS联合CDKN2A或TP53任一的缺失,不足以驱动人胰腺祖细胞发生PDAC转化。
3.KRAS、CDKN2A和TP53联合突变在移植后驱动PanIN和PDAC
患者中PDAC的进展以多个TSG的累积性改变为标志[5,6]。因此,我们使用共表达靶向CDKN2A和TP53 sgRNA的慢病毒载体生成了KCPPO。随后通过递送SMAD4 sgRNA至KCPPO并筛选TGF-β耐药克隆,引入了SMAD4缺失(图S2C)[29]。通过CRISPR测序和Western blot证实了TSG敲除成功(图2B,S2D,E)。
与注射K、KC或KPPO的小鼠无肿瘤形成相反,注射了传代匹配的KCPPO的22/23只小鼠出现了胰腺病变。组织病理学分析证实,镜下腺样病变类似于患者中观察到的PanIN(3/23)和中分化PDAC(19/23)(图2C)。KCPs异种移植物显示出更具侵袭性的肿瘤生长和广泛的促结缔组织增生反应(图2C)。在所有评估的KCPs病变中,中低分化PDAC占主导地位(13/26),未检测到PanIN(0/26),表明疾病快速进展(图2C,表S1)。
免疫组化(IHC)分析进一步确认了肿瘤身份。CK19染色显示KCP和KCPs异种移植物中存在肿瘤细胞(图2D,E,左两图),这些细胞E-cadherin+、高度增殖(Ki67+)且GFP+(来自GFP-KRASG12V融合蛋白),证实其来源于改造后的PO(图S2H-J)。阿尔辛蓝染色显示,KCP和KCPs肿瘤的腺样区域存在丰富的酸性粘蛋白,而在KCPs肿瘤中缺乏腺样结构的低分化区域染色减少(图2D,E),这与向更高肿瘤级别的进展一致[30]。对所有异种移植物切片的检查未发现畸胎瘤形成——这是hPSC疾病建模和细胞治疗中已知的混杂因素[31,32],可能干扰肿瘤特异性组织病理学表征。
因此,异种移植物组织病理学忠实地再现了与人类遗传进展一致的进行性PDAC发展(图2F)[5-7]。值得注意的是,KRAS、CDKN2A和TP53的联合突变(常见于高级别PanIN和患者PDAC中[5,8,9,33])赋予了PO致瘤能力。由此产生的异种移植物缺乏SMAD4缺失(KCPs),表现出更高级别的组织学和更晚期的分子特征。因此,我们的模型重现了患者PDAC中的累积遗传进展,其中KRAS、CDKN2A和TP53的突变是启动肿瘤发生的必要和充分条件(KCP),而额外的SMAD4缺失则促进肿瘤进展为更高级别的PDAC(KCPs)。
4.KCP和KCPs PO重现人PDAC转化和进展的转录组特征
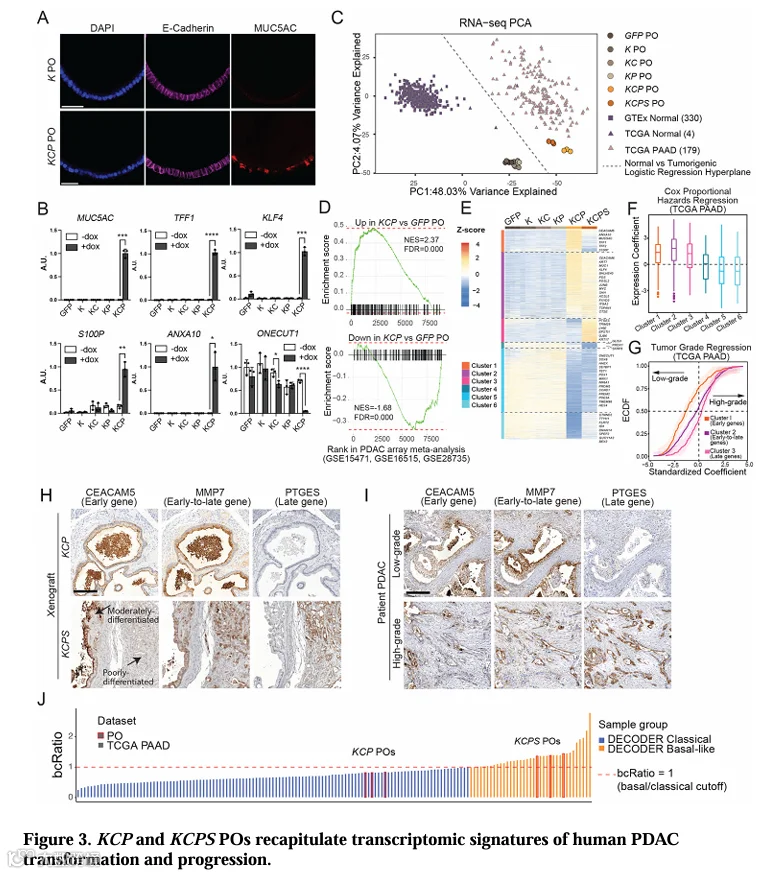
为了表征KCP和KCPs PO的分子特征,我们进行了RNA-seq分析。与非致瘤性K、KC和KPPO相比,KCP和KCPs PO在形态上表现出明显的上皮-间质转化(EMT)特征(图3A)。RT-qPCR分析显示,在KCP和KCPs PO中,多种恶性标志物(MUC5AC、CEACAM5、CEACAM6、TFF1、S100P、MMP7和KRT19)显著上调(图3B)。
为了将PO转录组与临床样本进行关联,我们将PO RNA-seq数据投射到由TCGA-PAAD和GTEx样本定义的PCA空间中。KCP和KCPs PO聚集在TCGA肿瘤样本附近,远离GTEx正常组织(图3C)。GSEA显示,在多个独立患者PDAC队列中,与PO转化相关的基因特征显著富集(图3D)。这些数据表明,KCP和KCPs PO的转录组特征与患者PDAC高度相似。
对PO RNA-seq数据中前3000个变异基因进行无监督聚类,鉴定出6个基因簇(图3E)。通过Cox回归分析和逻辑回归分析,这些基因簇与患者生存率和肿瘤级别相关联(图3F,G)。我们将在KCP向KCPs进展过程中表达下降的簇命名为“早期基因”,表达上升的簇命名为“晚期基因”,而表达先升后降或呈现中间模式的簇命名为“早期-晚期基因”(图3E,G)。IHC验证了这些基因在异种移植物和患者PDAC中的表达模式(图3H,I)。
DECODER分析[43]显示,KCP PO被分类为经典型(bcRatio<1),而KCPs PO被分类为基底样型(bcRatio>1)(图3J),与Moffitt PDA亚型分类一致[41]。这些分子分类结果表明,KCP和KCPs PO分别对应于早期转化和进展期PDAC。
5.染色质可及性分析揭示了PDAC发展过程中细胞可塑性的两个核心组成部分
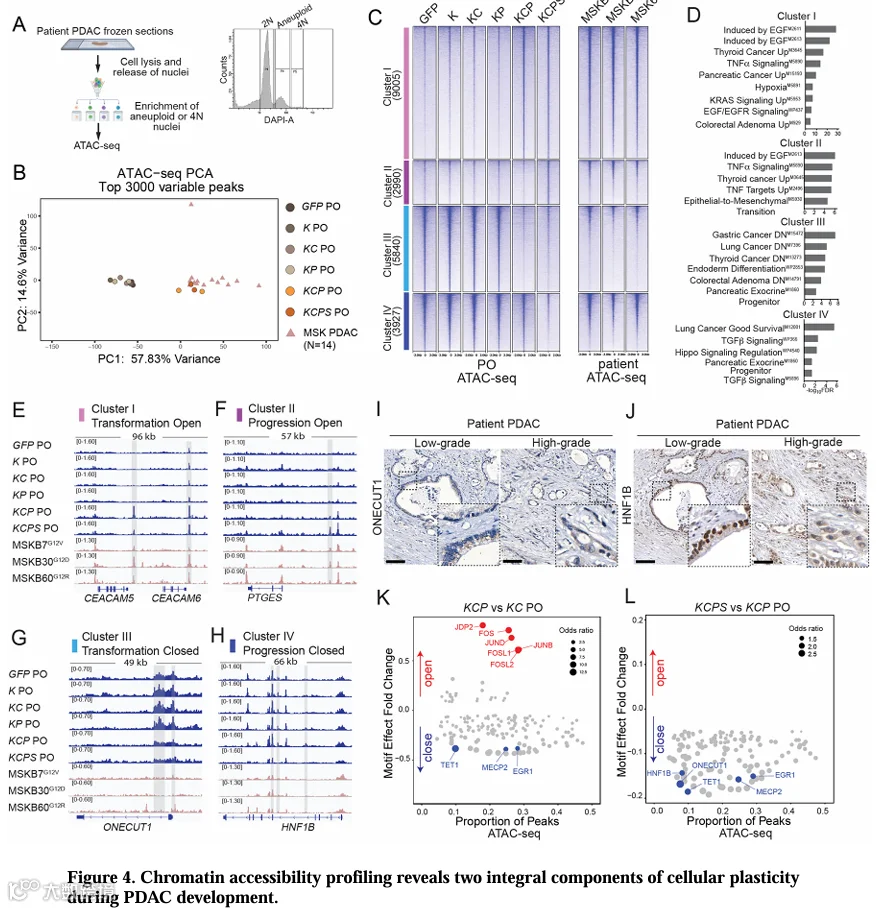
为了探索驱动PDAC恶性转化和进展的分子机制,我们对PO进行了ATAC-seq分析。为了评估患者样本中的表观遗传变化,我们还建立了一种从速冻和OCT包埋的患者肿瘤中分离非整倍体/多倍体肿瘤核的方法,以克服PDAC标本中肿瘤细胞含量低的问题(图4A,表S5)。与我们的RNA-seq结果一致,KCP和KCPs PO的染色质可及性谱与大多数患者PDAC样本聚类,但与非致瘤性的K、KC和KPPO不同(图4B)。
对21,762个可变ATAC-seq区域的聚类鉴定出四组基于PDAC发展过程中可及性动态变化的区域,并在患者样本中得到证实(图4C,表S6)。簇I区域(9,005个区域,转化开放)在KCP和KCPs PO中可及性增加,与KRAS、EGFR和TNF-α信号通路相关,并富集了MSigDB中内胚层来源恶性肿瘤相关的基因集(图4D)。这些区域包括PDAC标志物如CEA/CAM5/6、MMP7、ITGA2、MUC5AC和KLF4的调控元件,在致瘤性PO和患者PDAC中可及性升高(图4E,S4A)。簇II区域(2,990个区域,进展开放)在KCPs PO中获得特异性可及性,包括与晚期基因PTGES和EPSTI相关的位点,后者被确定为PDAC的不良预后标志物(图4F,S4B,E,F)。
簇III区域(5,840个区域,转化关闭)在KCP和KCPs PO中可及性降低,这些区域与内胚层分化相关基因有关,包括胰腺谱系转录因子ONECUT1、NR5A2、PDX1,在患者PDAC中也观察到类似的可及性丧失(图4C,D,G,S4C)。簇IV区域(3,927个区域,进展关闭)表现出KCPs特异性的可及性丧失,包括与胰腺谱系转录因子HNF1B、MNX1、HHEX以及TGFβ信号组分TGFBR2相关的区域(图4C,D,H,S4D)。胰腺谱系转录因子与簇III和IV区域的显著关联表明,PDAC的启动和进展涉及胰腺谱系程序的丢失。支持这一观点的是,来自hPSC胰腺分化[45]的ATAC-seq数据显示,胰腺特化涉及簇III/IV区域的开放和簇I/II区域的关闭(图S4G)。
簇III和IV区域可及性的逐步丧失与PDAC发展的不同阶段相关。因此,我们通过邻近性检测了与这些区域相关的转录因子的表达。IHC染色检测到,与正常导管相比,KCP异种移植物中ONECUT1信号在肿瘤上皮中减少,而在KCPs异种移植物中信号消失(图S4K,L)。类似地,患者PDAC中,无论肿瘤级别如何,核内ONECUT1染色均缺失(图4I),表明ONECUT1在早期恶性转化中就已丢失,这与我们的PO和TCGA PAAD转录组数据以及既往研究一致[46,47](图3B,S4H,I)。相比之下,HNF1B信号在KCP异种移植物中与正常导管相当,但在KCPs异种移植物中特异性地在低分化成分中减少(图S4M,N)。患者样本显示,低级别PDAC中核HNF1B染色强,而在高级别肿瘤中减弱(图4J,S4J)。因此,我们的模型揭示了在PDAC转化和进展过程中胰腺谱系程序进行性表观遗传抑制。特别是,HNF1B表达降低与癌细胞向低分化状态进展相吻合,这与患者IHC和临床报告一致,即核HNF1B染色缺失与部分PDAC患者术后预后不良相关[48]。总之,这些结果表明我们的模型重现了人PDAC的关键分子特征。
6.致癌性AP-1在早期PDAC转化过程中驱动谱系可塑性
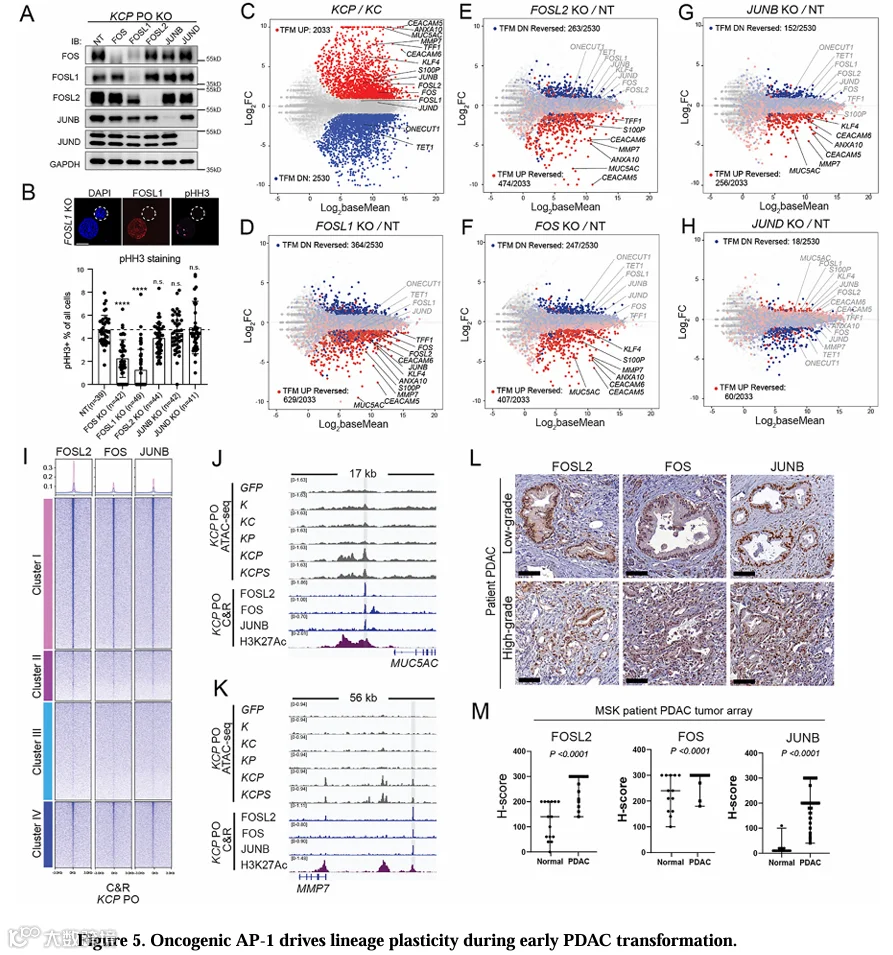
PDAC发展过程中染色质可及性的改变表明两个协调进行的过程:与跨谱系/恶性程序相关的区域获得可及性,以及与胰腺谱系程序相关的区域失去可及性(图4C)。AP-1基序在KCPPO恶性转化过程中获得可及性的区域中高度富集(图4K,S4O,P)。为了鉴定驱动这一过程的关键AP-1因子,我们优先选择了在KCPPO中高表达且基序富集的候选因子,并进行Cas9介导的扰动。由于FOSL1在KRAS驱动的PDAC肿瘤发生中具有不可或缺的作用[19,49,50],我们将其作为阳性对照。在KCPPO中递送sgRNA后4周,通过CRISPR测序和Western blot证实了高效基因扰动(图5A,S5A)。FOSL1缺失导致强烈的增殖缺陷,pHH3+细胞显著减少,验证了该方法的敏感性。类似地,FOS缺失也减少了KCPPO的增殖(图5B,S5A)。
为了评估单个AP-1因子对转录组的影响,我们检查了通过比较KCP与KCPO鉴定出的转化相关上调基因(TFM UP,n=2,033)和下调基因(TFM DN,n=2,530)(图5C)。正如预期,与转导非靶向sgRNA的KCPPO相比,FOSL1缺失降低了恶性特征的幅度(图5D)。KCPPO中FOSL2、FOS和JUNB的缺失导致类似的恶性特征减弱,而JUND缺失的影响最小(图5E-H,表S7)。RT-qPCR证实,在KCPPO中敲低FOSL1、FOSL2、FOS和JUNB后,恶性标志物MUC5AC、CEACAM5、ANXA10、MMP7显著下调,而S100P和KLF4的水平表现出基因型特异性下调。这些基因在JUND缺失的KCPPO中均未显示下调(图S5B)。通过免疫荧光验证了FOSL2、FOS和JUNB缺失的KCPPO中MUC5AC表达降低,证实了这些AP-1因子在PDAC中的致癌作用(图S5C-E)。
为了探索致癌性AP-1对恶性特征的机制调控,我们在KCPPO中进行了CUT&RUN(C&R)实验。FOSL2、FOS和JUNB在簇I(转化开放)区域显示出富集结合,其中主要检测到AP-1基序(图5I,S5F)。在这些区域中,MUC5AC和MMP7的调控元件被致癌性AP-1因子结合,并标记有活性组蛋白标志物H3K27ac(图5J,K),支持AP-1在PDAC表观遗传重编程中的功能作用。对FOSL2、FOS和JUNB的IHC显示,KCP和KCPs异种移植物中核染色富集(图S5H)。对MSKCC PDAC肿瘤阵列(PDAC:n=100,正常胰腺:n=13)的分析显示,这些AP-1因子在人PDAC中的表达显著升高(图5L,M),与PO和TCGA PAAD表达数据一致(图S5G,I)。值得注意的是,与其致癌作用一致,FOSL2的表达与患者不良预后相关,这与JUND的无预后相关性形成对比(图S5J)。总之,我们的数据表明,在PDAC转化过程中,多个AP-1因子(包括FOSL2、FOS、JUNB和FOSL1)协同作用,驱动与恶性特征相关的表观遗传重编程。
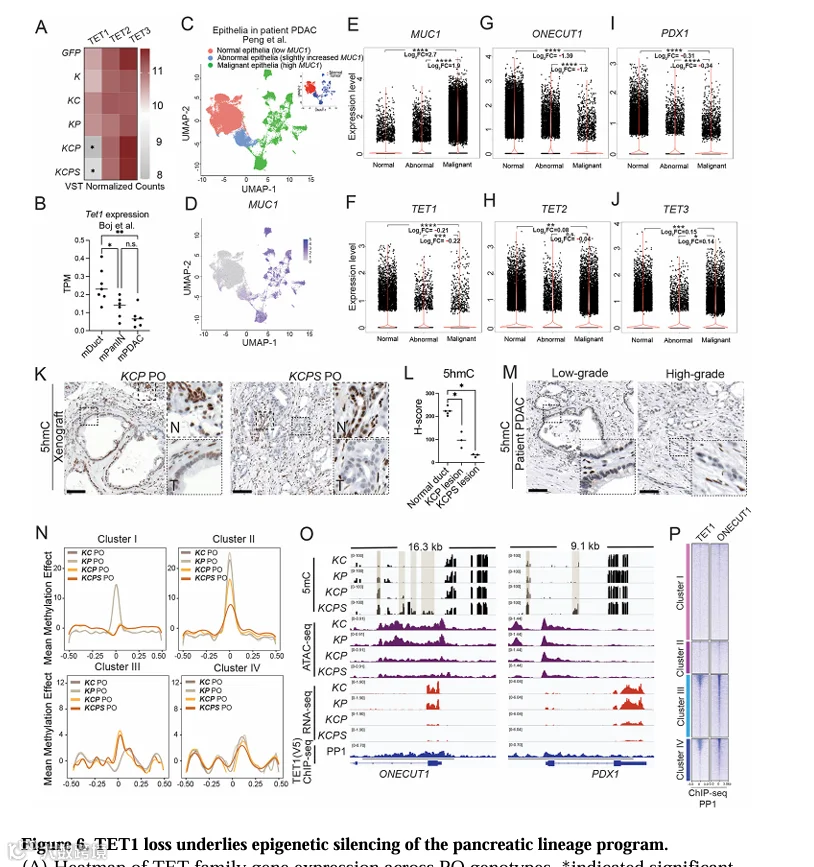
鉴于簇III和IV区域可及性丧失与胰腺谱系转录因子的关联(图4C,D,G,H),我们研究了PDAC转化过程中这些区域表观遗传沉默的机制。DNA甲基化在调节谱系特异性转录因子中起关键作用。我们首先检查了TET家族双加氧酶的表达,该家族催化5mC向5hmC的转化,是活性DNA去甲基化的关键步骤[52,53]。在KCP和KCPs PO中,TET1的表达显著降低,而TET2和TET3的表达变化较小(图6A)。类似地,在小鼠PDAC类器官中,Tet1的表达在PanIN和PDAC中相对于正常导管降低(图6B)。
为了探究TET1在PDAC中的临床相关性,我们分析了患者PDAC的单细胞RNA-seq数据[57]。随着上皮细胞从正常(MUC1低)向异常再到恶性(MUC1高)状态转变,TET1的表达逐渐降低(图6C-E)。相反,已知受TET1调控的DNA甲基转移酶DNMT1和维持CpG甲基化的UHRF1在恶性细胞中表达升高(图6F,G)。我们进一步评估了5hmC水平——TET活性的标志物。在KCP异种移植物中,与邻近正常导管相比,肿瘤上皮中5hmC信号降低,而在KCPs异种移植物中几乎完全消失(图6K,L)。在患者PDAC中,低级别肿瘤显示残留的5hmC染色,而高级别肿瘤中5hmC基本缺失(图6M)。这些数据表明,在PDAC转化过程中TET1受到抑制,导致5hmC丢失和DNA高甲基化。
为了直接评估DNA甲基化变化,我们对KCP和KCPs PO进行了EPIC甲基捕获测序。在簇III和IV区域(在转化和进展过程中可及性丧失)中,我们观察到KCP和KCPs PO相对于非致瘤性KC和KPPO出现高甲基化(图6N)。有趣的是,这些高甲基化区域富集了TET1结合基序(图6O)。在PP1细胞中进行的TET1 ChIP-seq证实,TET1结合在簇III和IV区域的启动子和增强子上(图6P)。此外,ONECUT1(胰腺谱系转录因子)也在这些区域结合(图6P)。这些发现表明,TET1缺失导致了PDAC转化和进展过程中胰腺谱系程序关键转录因子的表观遗传沉默。
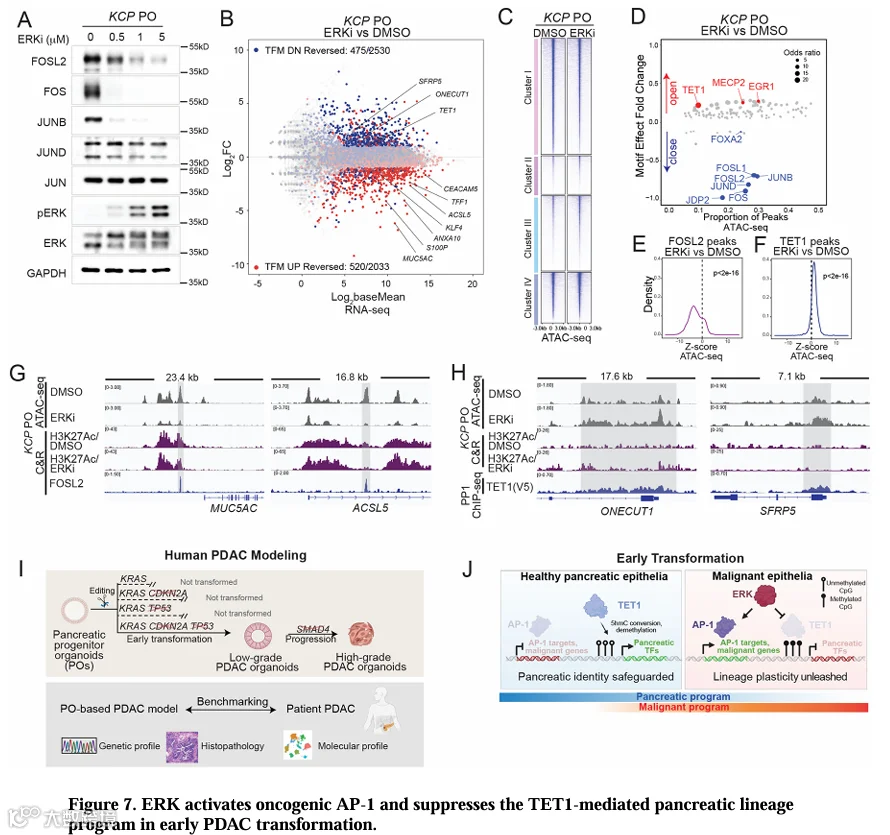
8.ERK在早期PDAC转化中激活致癌性AP-1并抑制TET1介导的胰腺谱系程序
MEK/ERK通路是PDAC和其他内胚层癌症中致癌性KRAS的关键下游效应器[61,62]。为了探究致癌性ERK信号如何影响人PDAC肿瘤发生过程中的转录组和表观遗传重编程,我们用ERK抑制剂temuterkib处理KCPPO[63]。temuterkib急性处理4天未诱导立即细胞死亡,但显著降低了FOS、FOSL2和JUNB的蛋白水平(图7A,S7A-C)。在整体水平上,ERK抑制部分逆转了KCPPO的恶性特征(图5C,7B)。在2,033个TFM UP基因(KCP与KC相比上调)中,有520个被ERK抑制下调,包括恶性标志物MUC5AC、TFF1、ANAX10和S100P,通过RT-qPCR验证。相反,2,530个TFM DN基因中有475个上调,包括ONECUT1和TET1(图7B,S7D,表S7)。
检查ERK抑制后KCPPO的染色质可及性景观,我们观察到簇I(转化开放)区域的可及性显著丧失,以及簇III(转化关闭)区域的重新开放(Wilcoxon符号秩检验,p<2×10^{-16},图7C,S7E,F)。对ERK抑制剂与DMSO处理的KCPPO进行基序分析表明,获得可及性的染色质区域富集TET1基序,而失去可及性的区域富集AP-1基序(图7D),表明ERK信号反向调节这些相关程序。为了测试ERK抑制是否影响真实TF结合位点的染色质,我们检查了KCPPO中致癌性AP-1(FOSL2、FOS、JUNB)结合区域以及PP1细胞中TET1或ONECUT1结合区域的染色质可及性(图5I,6P)。在全基因组范围内,ERK抑制导致AP-1结合区域的可及性降低,而TET1和ONECUT1结合区域的可及性增加(Wilcoxon符号秩检验,p<2×10^{-16},图7E,F,S7I-K),与其在KCPPO中ERK抑制后的表达模式一致。此外,与簇I中未结合区域相比,致癌性AP-1结合区域的可及性丧失更为显著(图S7L-N),而簇III中TET1结合区域的可及性增加更为突出(图S7O)。ONECUT1结合区域在簇III区域中未显示优先开放(图S7P)。例如,ERK抑制抑制了与MUC5AC和ACSL5相关的FOSL2结合的簇I区域(图7G),并恢复了与ONECUT1和SFRP5相关的TET1结合的簇III区域的可及性(图7H)。这些发现表明,ERK信号在KCPPO中激活致癌性AP-1因子,同时抑制TET1,进而抑制胰腺谱系程序。总之,这些协调进行的过程驱动了人PDAC恶性转化过程中的细胞可塑性。
更多结果和补充图表:doi:10.1016/j.devcel.2026.04.012
长按二维码关注我们,用最短的时间和最高的效率学习更多生信思路!
扫描上方二维码或登录平台官网后添加CNSknowall客服微信咨询!官网地址:https://cnsknowall.com
CNSknowall:24年最新问世的遥遥领先的颠覆性科研数据(0代码生信+统计学)分析平台,同时含有机制图模块(原创3000多素材和机制图模板)+AI一键生成高质量比国自然标书初稿+汉化版Pubmed融合Deepseek高效筛选目标文献同时一键提炼全文核心创新点+SCI文献例句/语料检索模块+全文翻译+文献求助+图片查重+期刊查询+OPenAI官方GPT接口,>500款CNS级别图表皆可一秒内一键出图,登录即秒变数据分析大神,体验前所未有的便捷数据分析之旅,开启科研天骄之路!
可向下滑动发掘更多科研秘籍!