炎炎的夏日与盛夏的蝉鸣声中,小记者(迫真!ps:实则是凄惨的科研打工人一枚!)不知多少次伏案桌前,在键盘上激情敲打着字符,键盘声伴着不断下落的头发被坚定地敲击而下。(ps:小记者不禁心中呐喊:这属实是用秃头的代价来进行科研啊)
科研的路上总是坎坷的!如果屏幕前的小伙伴也有科研的烦恼,那就来生信日报公众号看看吧!
小记者这两天在PubMed里发现了一篇题目中规中矩,选题方向新颖且结合了当下的人工智能热潮的4分+的文章。在细细品读研究后,顿时觉得自己又发现了生信文章中高分期刊的秘诀。接下来让小记者向各位小伙伴分享这篇文章是怎样做到的吧!
小记者发现作者通过开发一种新的深度学习框架LPI-DLDN,该研究成功地识别了潜在的lncRNA-蛋白质相互作用,并在性能和准确性方面优于六种现有的LPI预测方法。这项研究为深入理解lncRNA的生物学功能和分子机制提供了新的工具和方法。文章的篇幅正常,且有九张图,但胜在逻辑分析清晰,数据调理有当,简单明了,创新有佳!(ps:想要学习如何形成独属于自己的科研创新思维,就快来找小记者聊聊吧!

题目:基于双网络神经结构深度学习的lncrna -蛋白相互作用研究
杂志:IEEE/ACM Trans Comput Biol Bioinform
影响因子:IF=4.5
发表时间:2022年12月
后台回复“999”领取文献
lncRNA(长链非编码RNA)是一类长度超过200个核苷酸的RNA分子,它们不编码蛋白质,但在细胞内发挥着重要的调控作用。近年来,越来越多的研究表明,lncRNA通过与RNA结合蛋白相互作用来调节细胞过程,从而发挥其功能。因此,识别潜在的lncRNA-蛋白质相互作用对于揭示lncRNA的生物学功能和分子机制非常重要。然而,目前的实验方法需要大量的时间和资源,因此需要开发新的计算方法来预测lncRNA-蛋白质相互作用。这篇文章的研究背景就是在这个背景下,通过开发一种新的深度学习框架来预测lncRNA-蛋白质相互作用。
数据集/队列 |
数据库 |
数据类型 |
详细信息 |
Datasets 1, 2, and 3 |
NPInter v3.0 |
基因数据 |
人类和小鼠的lncRNA-蛋白质相互作用数据,共计16,969个相互作用对 |
Datasets 4 and 5 |
RPI2241 |
基因数据 |
人类和小鼠的RNA-蛋白质相互作用数据,共计2,241个相互作用对 |
作者使用了两个公开的数据集,分别是NPInter v3.0和RPI2241。研究对这些数据进行了预处理,去除了无效数据和异常值。(ps:想要学习如何选择合适的统计学方法?那快来生信日报找小记者聊聊吧!)使用了多种特征来描述lncRNA和蛋白质的序列和结构信息,包括序列特征、结构特征和进化特征。这些特征是通过多种计算方法和工具来提取的,包括RNAfold、RNAsnp、PSSM等。使用了一种新的深度学习框架LPI-DLDN来预测lncRNA-蛋白质相互作用。该框架包括两个部分:特征提取和分类器。特征提取部分使用了多层卷积神经网络(CNN)和长短时记忆网络(LSTM)来提取lncRNA和蛋白质的特征。分类器部分使用了多层感知机(MLP)来预测lncRNA-蛋白质相互作用的概率。为了提高模型的性能,研究还使用了一种双网络结构,即将lncRNA和蛋白质分别输入到两个不同的CNN-LSTM网络中,然后将它们的输出连接起来,再输入到MLP中进行分类。使用了多种指标来评估模型的性能,包括准确率、召回率、F1值、AUC值等。为了验证模型的泛化能力,研究还进行了交叉验证和外部测试。此外,研究还与其他预测模型进行了比较,证明了LPI-DLDN的优越性。
作者利用六项指标来评估提出的LPI-DLDN框架的性能:精确度、召回率、准确度、F1-分数、ROC曲线下面积(AUC)和精确度-召回率曲线下面积(AUPR)。这六个指标在统计上是一致的标准。精确度、召回率、准确率、F1-分数、AUC和AUPR越高,表示性能越好。对比实验重复20次,最终性能由20次迭代结果的平均值得出。
在基于深度学习的预测模型中,关联概率大于0.5的LPI被归为正类,小于0.5的LPI被归为负类。在本研究中,作者采用函数 sklearn. metrics.roc_curve函数来生成阈值数组。该数组中的阈值用于计算6个测量值,最后计算平均性能。
作者将Pyfeat用于提取lncRNA特征,参数设置如下:kGap = 5, kTuple = 3, opti-mumDataset = 0, pseudoKNC = 1, zCurve = 1, gcContent = 1, cumulativeSkew = 1, atgcRatio = 1, monoMono = 1, monoDi = 1, monoTri = 1, diMono = 1, diDi = 1, diTri = 1, triMono = 1, and triDi = 1. BioTriangle中的所有特征都用于表示蛋白质。LPI-HeteSim 中的参数设置为 Zhou 等人提供的默认值。其他6个LPI预测模型的参数设置为模型通过网格搜索获得最佳性能的相应值(表2)。
作者采用网格搜索,发现当d ¼ 100时,LPIDLDN获得了更好的性能。因此,作者提取了两个100维向量来表示lncRNA和蛋白质特征。为了测量LPI-DLDN的性能,作者进行了4次不同的5倍交叉验证(CV)。
1)对lncRNA的5倍交叉验证(CV1):Y中的随机行被屏蔽用于测试,即每轮选择80%的lncRNA作为训练集,剩余的20%作为测试集。
2)蛋白质的5倍CV(CV2):Y中的随机列被屏蔽用于测试,即每轮选择80%的蛋白质作为训练集,其余20%作为测试集。
3) lncRNA 蛋白对的 5 倍 CV(CV3):在每一轮中屏蔽 Y 中随机的 lncRNA 蛋白对进行测试,即选取 80%的 lncRNA 蛋白对作为训练集,剩余 20%作为测试集;4) 独立 lncRNA 和独立蛋白的 5 倍 CV(CV4)[48]:在每一轮中屏蔽 Y 中随机的 lncRNA 蛋白对进行测试,即选取 80%的 lncRNA 蛋白对作为训练集,剩余 20%作为测试集。
4)独立lncRNA和独立蛋白质的5倍CV(CV4)[48]:首先,随机选择20%的lncRNA和20%的蛋白质组成 "节点测试集"。第二,剩余的节点(lncRNA或蛋白质)被视为 "节点训练集"。第三,从 "节点训练集 "中的节点到 "节点测试集 "中的节点的所有连接边被丢弃并排除在分析之外。
最后,一个分类器只对节点训练集中的边缘进行训练,以预测节点测试集中的边缘。
(1)新的(未知的)lncRNA(即不与任何蛋白质相互作用的lncRNA);
(2)新的蛋白质(即不与任何lncRNA相互作用的蛋白质);
(4)新的独立的lncRNA和独立的蛋白质的LPI预测。
此外,从未标明的lncRNA-蛋白质对中随机选择阴性LPI。所选阴性样本与阳性样本的比例为1,即阴性LPI的数量与已知LPI的数量相同。
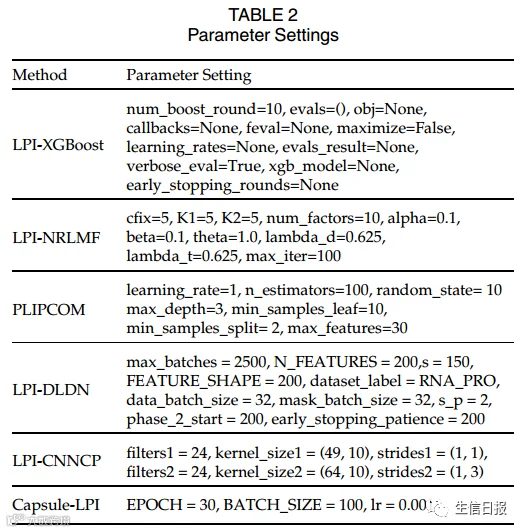
表2 化疗对所有种族的益处可以用随机森林生存来模拟
作者将提出的LPI-DLDN方法与6种最先进的LPI预测方法,即LPI-XGBoost、LPIHeteSim、LPI-NRLMF、PLIPCOM、LPI-CNNCP和Capsule-LPI进行比较,以评估LPI-DLDN的预测能力和鲁棒性。图2、图3、图4和图5分别显示了7种LPI预测方法在5个数据集上4种不同交叉验证下的ROC曲线和PR曲线。
LPI-DLDN在5个数据集上获得了最高的平均精度、F1-score和AUC,明显优于LPI-XGBoost、LPI-HeteSim、LPI-NRLMF、PLIPCOM、LPI-CNNCP和Capsule-LPI。虽然LPI-DLDN计算的平均召回率、准确率和AUPR分别略低于Capsule-LPI、LPI-XGBoost和LPI-HeteSim计算的平均召回率、准确率和AUPR,但差异很小,可以忽略不计。
例如,Capsule-LPI计算的平均召回率为0.7722,而LPI-DLDN得到的值为0.7687,小于CapsuleLPI的0.46%。LPI-XGBoost计算的平均精度为0.8199,而LPI-DLDN的值为0.8165,仅小于LPI-XGBoost的0.40%。LPI-HeteSim得到的平均AUPR为0.8185,而LPI-DLDN得到的平均AUPR为0.8150,仅小于0.43%。
图2展示了CV1下7种LPI预测模型在5个数据集上的ROC曲线和精度-召回(PR)曲线。LPI-XGBoost、LPI-HeteSim、LPI-NRLMF、PLIPCOM、LPI-CNNCP和Capsule-LPI是目前最先进的LPI预测方法,在新LPI识别中获得了优异的性能。LPI-DLDN要么明显优于6种竞争模型,要么差异很小。因此,LPI-DLDN在寻找与新的lncRNA相互作用的蛋白质方面是强大的。
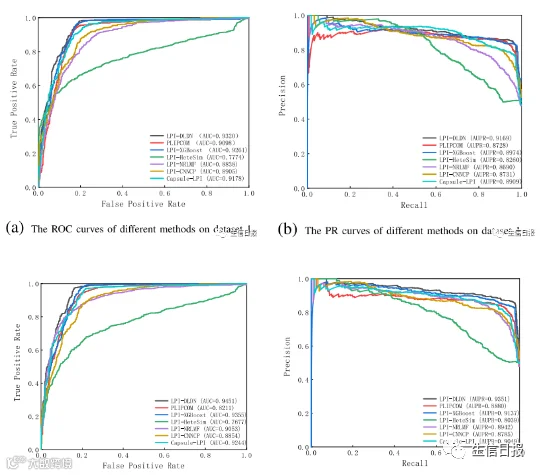
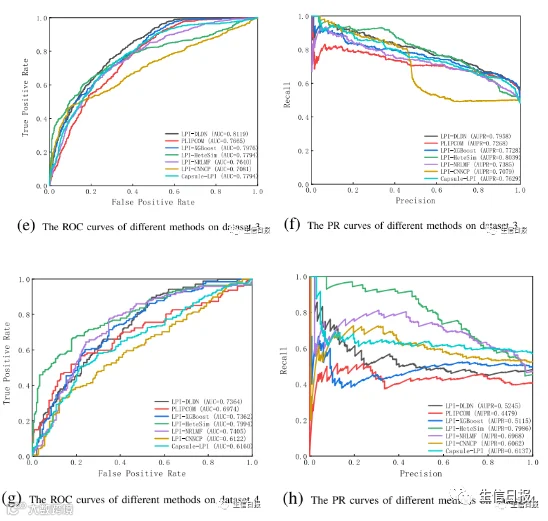
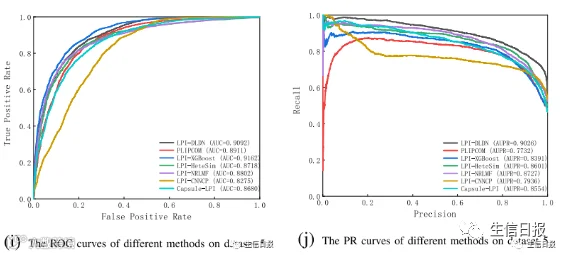
图2 不同方法在CV1下的ROC曲线和PR曲线
如表2所示,虽然LPI-HeteSim和LPI-NRLMF的平均性能略优于LPIDLDN,但这两个基于网络的LPI预测模型存在一个严重的不足:基于网络的模型无法找到孤lncRNA(或蛋白质)可能的相互作用信息。更重要的是,在大多数条件下,LPI-DLDN优于LPI-XGBoost和PLIPCOM这两种基于集合学习的LPI推断方法。特别是,AUPR是比其他五个指标更重要的衡量指标。LPI-DLDN的平均AUPR优于LPI-XGBoost和PLIPCOM。结果表明,LPI-DLDN可能是一种有效的监督学习方法,可用于识别与新蛋白质相关的潜在lncRNA。此外,LPI-DLDN优于LPI-CNNCP,后者是一种基于深度学习的LPI识别技术,再次证明了LPI-DLDN的优越性。
图3描述了CV2条件下7种LPI预测方法在5个数据集上的ROC曲线和textcolorredPR曲线。结果表明,在所有数据集上,LPI-DLDN在精度、召回率、F1-score、AUC和AUPR方面都明显优于其他六个LPI预测模型。
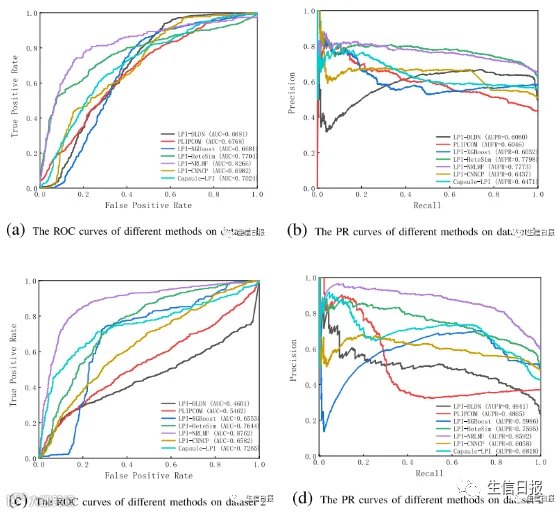
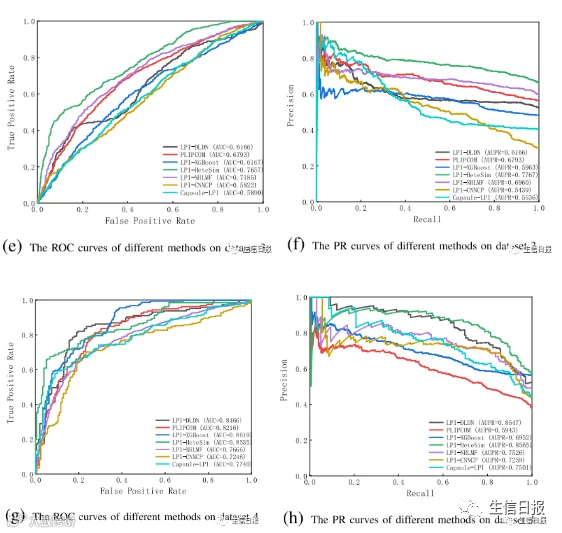
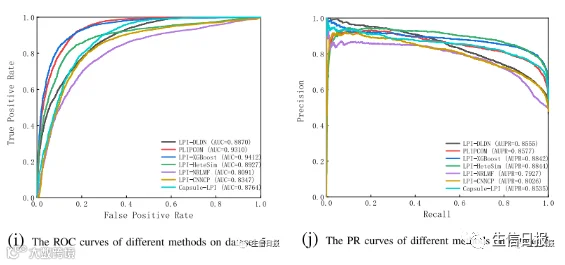 图3 不同方法在CV2下的ROC曲线和PR曲线
图3 不同方法在CV2下的ROC曲线和PR曲线
例如,LPI-DLDN计算出的最佳平均AUC值为0.9110,分别比LPI-XGBoost、LPI-HeteSim、LPI-NRLMF、PLIPCOM、LPI-CNNCP和Capsule-LPI好1.22%、11.27%、2.29%、2.65%、7.40%和0.95%。更重要的是,在AUPR指标上,LPIDLDN的平均AUPR为0.8984,比第二好的方法高1.46%,比第三好的方法高2.0%。
图4是7种LPI预测模型在CV3下5个数据集上的ROC曲线和PR曲线。结果表明LPI-DLDN具有强大的分类能力。因此,LPI-DLDN可用于在已知LPI的基础上发现lncRNA与蛋白质之间新的相互作用。
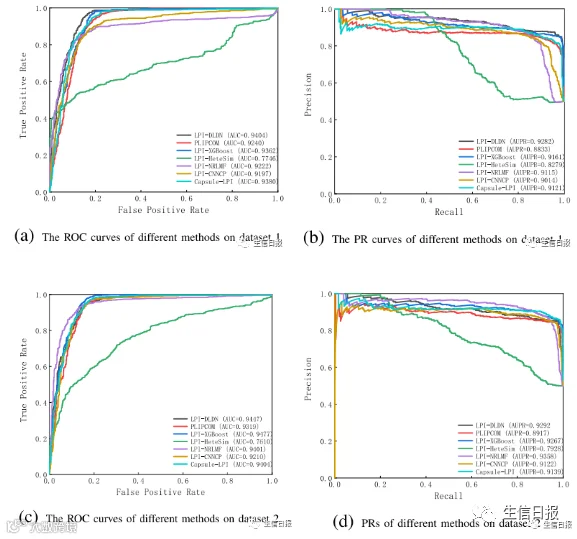
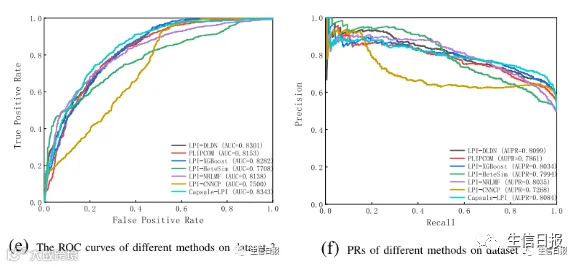
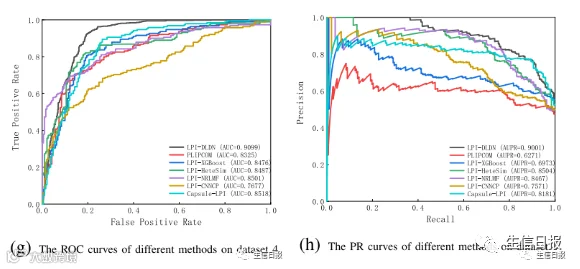
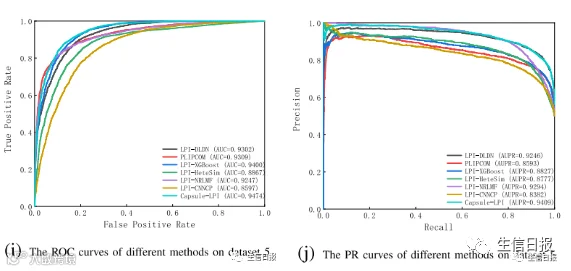
图4 不同方法在CV3下的ROC曲线和PR曲线
CV4能够确保在分析过程中不发生数据泄露,并确保训练模型没有通过连接到训练集中的另一个节点而看到测试集中的任何节点。在大多数条件下,LPI-DLDN明显优于LPI-XGBoost、LPIHeteSim、LPI-NRLMF、PLIPCOM和LPI-CNNCP。更重要的是,尽管Capsule-LPI计算出了更好的精度、召回率、准确率和F1-score,但LPI-DLDN在5个数据集上获得了最优的平均AUC和AUPR。与其他四个评价指标相比,AUC和AUPR是两个更具代表性的测量指标。LPI-DLDN计算出的最佳平均AUC为0.6527,比Capsule-LPI高3.7%,最佳AUPR为0.6372,比Capsule-LPI高0.78%。图5显示了7种LPI预测方法在CV4下5个数据集上的ROC曲线和PR曲线。
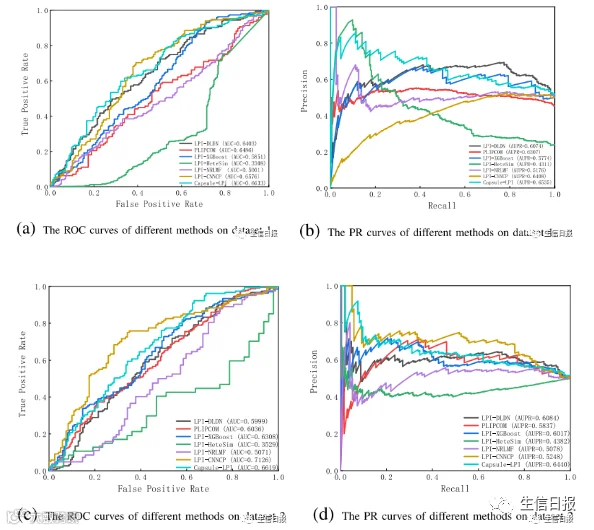
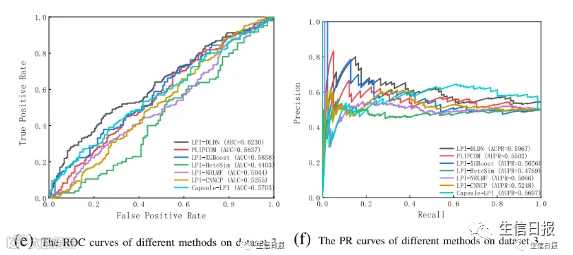
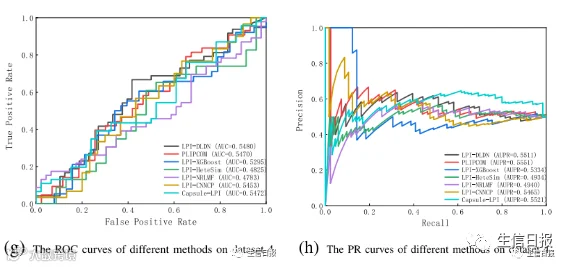
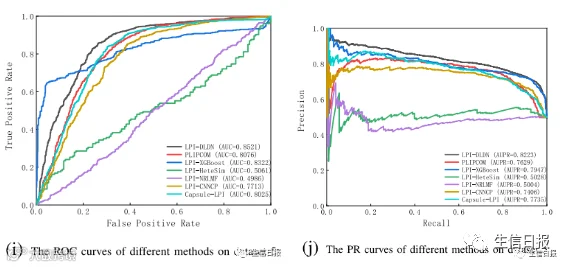
图5 不同方法在CV4下的ROC曲线和PR曲线
作者受Lan等人提供的数据分布描述的启发,研究了每个LPI网络中的度。本节以lncRNA为节点,分析了5个LPI网络。结果如图6所示。在数据集1-3中,节点的度分布非常不均匀。例如,在数据集1-3中,大多数节点的度数小于12。度数为2的节点数分别为360、340和378,在三个人类数据集中占了很大比例。在数据集4中,节点的度数分布相对均匀。在数据集5中,节点的度数分布比较均匀,度数为42的节点有603个。数据的不均匀性和不平衡性特征导致在数据未经筛选的情况下出现预测偏差。也就是说,预测结果会偏向于某一类。因此作者在训练集和测试集中选择相同数量的正样本和负样本,以减少预测偏差。
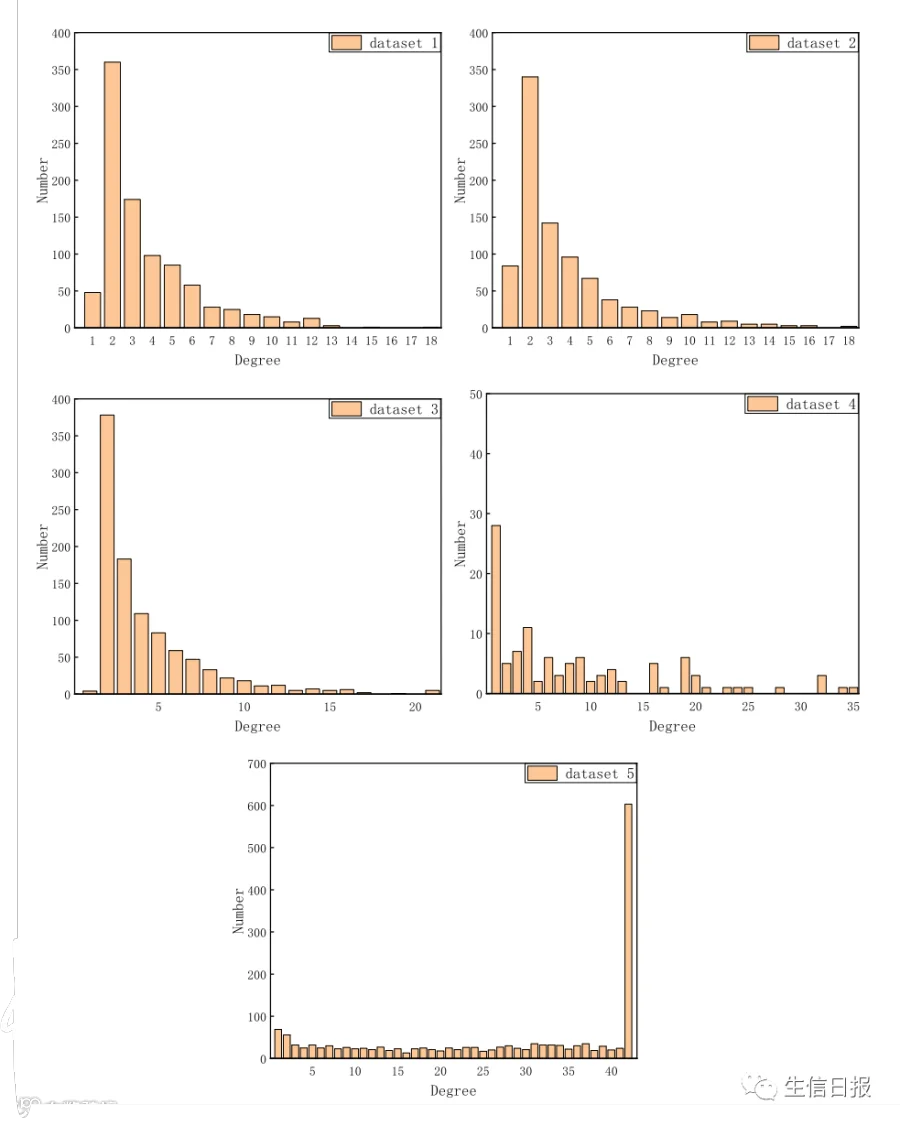 图6 五个LPI数据集的LPI网络度
图6 五个LPI数据集的LPI网络度
s表示从2d个LPI特征中选取的最高的s个特征,且s<2d。为了评估s对分类性能的影响,作者将其设置在(1,200)的范围内,间隔为25,并研究了LPI-DLDN在5个LPI数据集上的性能,在4个交叉验证下。结果如图7所示。从图7中可以发现,当特征个数s设置为150时,LPI-DLDN计算出了最优的AUC和AUPR。因此,作者选择s为150。
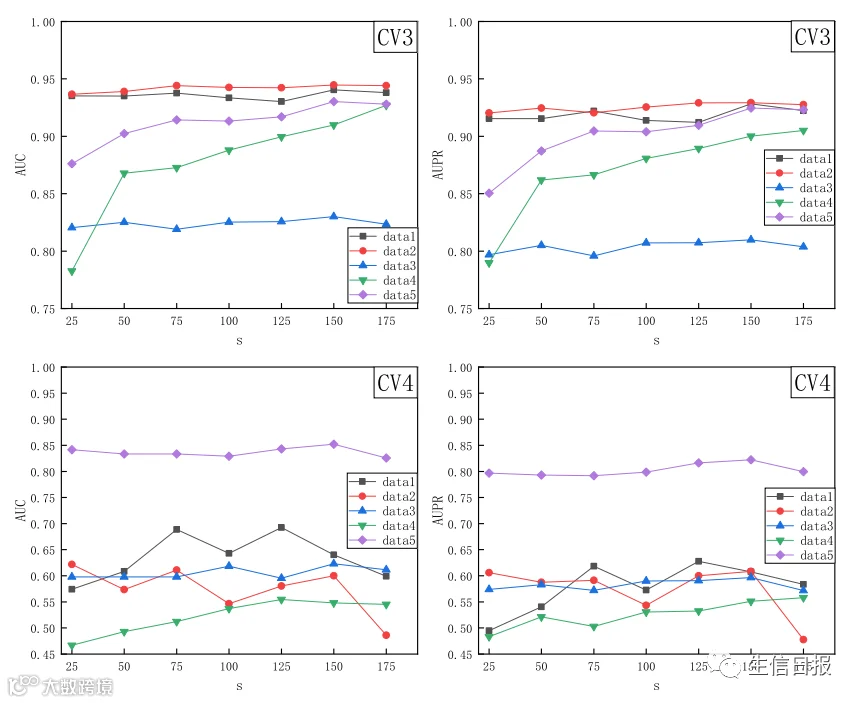 图7 AUCs 和 AUPRs基于四种交叉验证下的不同特征号
图7 AUCs 和 AUPRs基于四种交叉验证下的不同特征号
在LPI-DLDN框架中,作者使用由MLP网络和FIR网络组成的双网络结构来选择最佳特征并预测每个lncRNA蛋白质对的标签。为了比较双网络结构(LPI-DLDN)和MLP网络(LPI-MLP)的性能,作者研究了LPI-DLDN和LPI-MLP的AUC和AUPR值。结果如图8所示。从图8中可以看出,LPI-DLDN在四个交叉验证的五个LPI数据集上获得了比LPI-MLP更好的AUC和AUPR。结果表明,双网架构明显优于MLP网,体现了深度学习的最优分类能力。因此,在LPI分类中使用双网结构是非常有必要的。
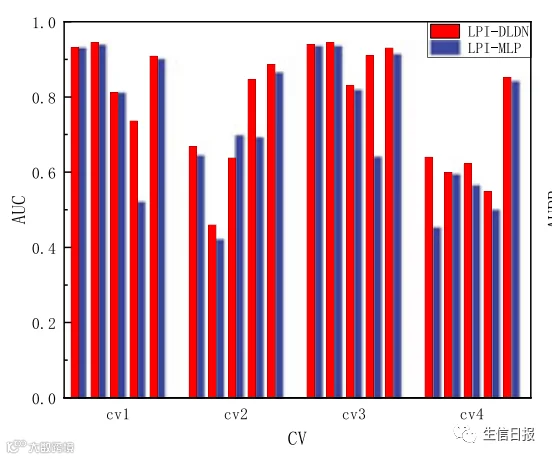
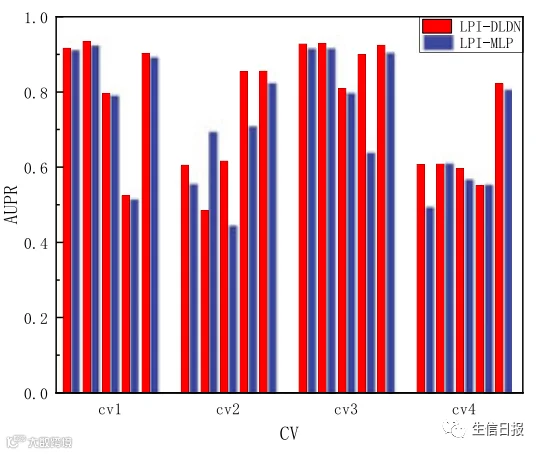
图8 LPI-DLDN和LPI-MLP的AUCs和AUPRs
在最后一节中,LPI-DLDN的性能得到了验证。作者进一步发现潜在的LPIs,特别是新的lncRNA(或蛋白质)可能的蛋白质(或lncRNAs)。
lncRNA FGD5反义RNA 1(FGD5-AS1)对多种人类癌症有重要影响。例如,FGD5AS1通过抑制结直肠癌细胞的迁移、侵袭和增殖,加速细胞凋亡,可作为结直肠癌的可能治疗靶点。通过与针对USP21的miR520b结合,它可以作为口腔鳞状细胞癌的诊断生物标志物。它可以通过hsa-miR-1533p/CITED2的下游表观遗传轴调控人类胃癌,并通过海绵状hsa-miR-107上调FGFRL1促进非小细胞肺癌的细胞增殖。
在数据集1-3中,FGD5-AS1(NONHSAT088370、n384228和NONHSAT088370)分别与6、6和8个蛋白相互作用。为了鉴定与FGD5-AS1相互作用的新蛋白,作者屏蔽了其所有的关联信息,并将其作为一个新的lncRNA。然后使用7种LPI鉴定方法鉴定与FGD5-AS1相关的潜在蛋白。实验重复10次,选出与FGD5-AS1相互作用的前5个预测蛋白。作者观察到,在数据集3中,O00425、Q9Y6M1和Q9NZI8被预测与FGD5AS1相互作用。尽管在数据集3中上述3个蛋白与FGD5-AS1之间的关系未知,但在数据集1中O00425已被证实与FGD5-AS1相互作用,在数据集1和2中Q9Y6M1和Q9NZI8已被报道与FGD5-AS1相互作用。更重要的是,在LPI-XGBoost、LPI-NRLMF、PLIPCOM、LPI-CNNCP和Capsule-LPI中,预测的与FGDAS1相互作用的前5个蛋白都有较高的排名。结果表明LPI-DLDN对新的lncRNA具有强大的相互作用预测能力。
7.2寻找与一种新蛋白质相互作用的潜在lncRNA 蛋白质
作者鉴定了可能与新蛋白相互作用的lncRNA。Q9H9G7是RNA介导的基因沉默所必需的。该蛋白与短RNA结合并抑制与之互补的mRNA的翻译。这一过程包括稳定干细胞中的小RNA衍生物以及依赖siRNA降解RNA聚合酶II转录的编码mRNA。同时,它仍然具有RNA切片活性。
在数据集1、2和3中,Q9H9G7分别与126、126和137个lncRNA相互作用。作者屏蔽了Q9H9G7的所有关联信息,并使用提出的LPI-DLDN方法来识别与该蛋白相互作用的潜在lncRNA。作者反复进行10次实验,得到所有lncRNA-蛋白质配对的平均关联得分。作者预测lncRNA n343060可能与Q9H9G7有关联,在数据集2上的排名为3。此外,在885个可能与Q9H9G7相关的lncRNA中,n343060与Q9H9G7之间的相互作用在其他6种LPI预测方法中的排名分别为18、207、322、2、820和738。结果表明,n343060可能与Q9H9G7存在相互作用,但这还有待进一步的实验验证。
7.3基于已知LPI寻找新LPI
作者基于LPI-DLDN进一步预测新的LPI。作者重复实验10次,计算数据集1-5上所有lncRNA-蛋白对的平均相互作用概率。图9显示了在5个数据集上预测的前50个LPIs,这些数据集包含已知的LPIs。图中,天蓝色实线和黑色虚线分别代表从LPI-DLDN得到的已知和未知LPI。深天蓝色圆圈和深橙色六边形分别代表关联信息已知和未知的lncRNA。绿色菱形表示蛋白质。
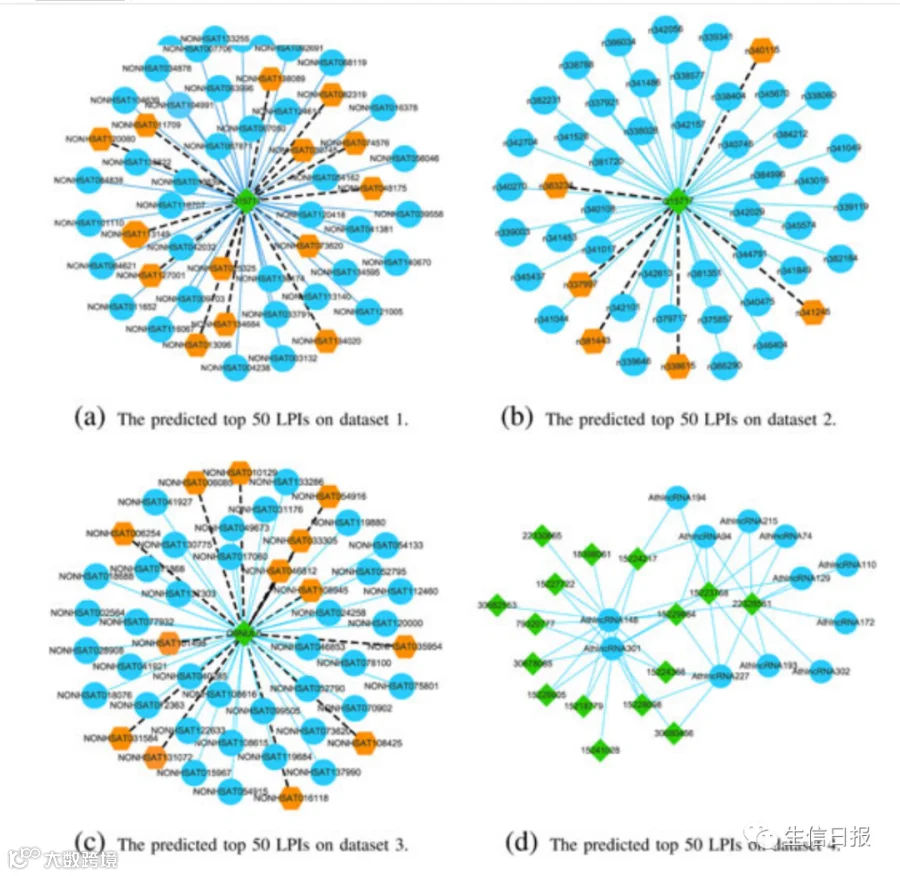
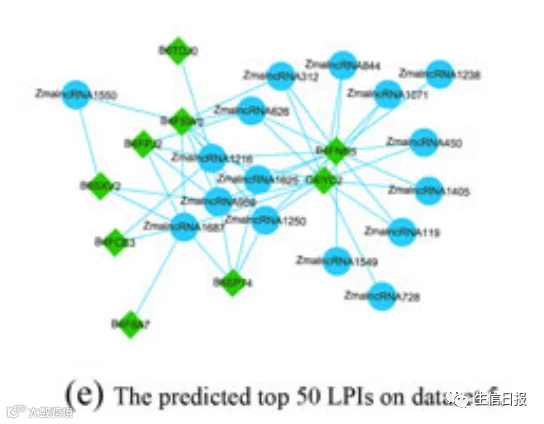
图9 在五个LPI数据集上预测的前50个LPI
NON-HSAT011709(RPI001_ 236932)与Q15717、n338615(RP11-439E19.10)和Q15717、NONHSAT006254(RP11-196G18. 22)和Q9NUL5、Athlnc RNA309(TCONS_00051077)和F4JLJ3、ZmalncRNA 1625和B8A305在5个数据集的未知lncRNA-蛋白配对中概率最高。在5个数据集中分别有55,165、74,340、26,730、3,815和71,568个lncRNA-蛋白质对。在所有的lncRNA-蛋白质对中,预测的5种相互作用分别为3、13、7、583和853。RP11-439E19.10被发现上调。该lncRNA可能通过与促炎细胞因子相互作用促进卵巢肿瘤的发生和发展。更重要的是,它可能与食管癌干细胞的放射敏感性有关,可作为食管鳞癌的新靶点。Q15717是一种RNA结合蛋白,有助于胚胎干细胞分化,调节p53/TP53的表达,介导CDKN2A的抗增殖活性,增加瘦素mRNA的稳定性。
在数据集2中,RP11-439E19.10被证实与Q13148、P35637和Q01844相互作用。Q13148调节与神经元存活相关的蛋白和编码神经退行性疾病蛋白的mRNA的剪接。它可以控制mRNA的稳定性,在维持昼夜节律周期性和线粒体平衡中发挥重要作用。它还参与正常骨骼肌的形成和再生[39]。P35637与多种细胞过程密切相关。该蛋白可结合自身的前mRNA并自动调节其表达。它在神经细胞的树突棘形成和稳定性、mRNA稳定性和突触平衡中发挥关键作用。
Q01844在肿瘤发生过程中发挥重要作用。该蛋白可能会干扰基因表达,协助融合蛋白靶基因的异常激活。Q15717与Q13148、P35637和Q01844具有相似的功能。根据 "关联罪 "原理,相似的lncRNA可能与相似的蛋白相互作用。更重要的是,在数据集1的所有55165个lncRNA-蛋白配对中,RP11-439E19.10和Q15717之间的相互作用被LPI-DLDN列为3。因此,作者推断RP11-439E19.10可能与Q15717具有紧密的联系。
此外,作者预测RP11-196G18.22可能与肺腺癌和邻近的正常样本密切相关。Q9NUL5可抑制来自病毒和细胞基因的多种mRNA的程序性-1核糖体框架转换(-1PRF)。该蛋白可导致翻译过早终止。它可以阻止 DENV RNA 的翻译,中断寨卡病毒的复制,限制丙型肝炎病毒的复制。作者推断RP11196G18.22可能与Q9NUL5相互作用,在所有26,730个lncRNA-蛋白配对中排名第7;然而,这一结论需要进一步验证。
这篇文章的科研方法比较全面和系统,包括数据预处理、特征提取、模型构建和模型评估等多个环节,每个环节都使用了多种方法和工具,(ps:想要学习如何选择合适的统计模型?那快来小记者公众号找小记者聊聊吧!)。使得研究具有较高的可靠性和可重复性。在创新性的方面,作者提出了一种新的深度学习模型LPI-DLDN,该模型采用双网络架构,由多层感知器(MLP)网络和有限脉冲响应(FIR)网络组成,用于识别潜在的长非编码RNA-蛋白质相互作用(PI)。相比于仅使用MLP网络的LPI-MLP模型,LPI-DLDN模型在五个LPI数据集上的AUC和AUPR值表现更好,证明了双网络架构的优越性和深度学习的最佳分类能力。作者引入了一种新的特征选择方法(ps:探索性的研究方法会更让审稿人眼前一亮喔!小记者愿意和你一起探讨创新性的研究思路!一起学习创新性的生信思路!),该方法基于FIR网络,可以选择最佳的LPI特征,并进一步提高LPI-DLDN模型的生成能力。同时,作者提出了一种新的LPI数据集,该数据集包含了大量的LPI数据,并且具有更好的数据质量和数据分布,可以用于评估和比较不同的LPI预测模型。通过对比实验,证明了LPI-DLDN模型在LPI预测任务中的优越性,也证明了FIR网络在特征选择和模型生成方面的有效性,为揭示lncRNA的生物学功能和分子机制提供了新的思路和方法。
小记者通过这篇文章也开阔了自己的科研思路(ps:来生信日报,和小记者一起打开生信的研究思路吧!),深度学习作为机器学习的一个分支,在生物信息学领域有着广泛的应用。通过深度学习,可以提高生物信息学数据的预测和分类能力,促进生物学研究的发展。特征选择是生物信息学研究的重要环节,可以提高模型的性能和泛化能力。通过特征选择,可以去除冗余和无关的特征,提取有用的特征,使得模型具有更好的表达能力和区分度。
这篇文章的科研方法比较全面和系统,作者提出了一种新的深度学习模型LPI-DLDN,该模型采用双网络架构,由多层感知器(MLP)网络和有限脉冲响应(FIR)网络组成,用于识别潜在的长非编码RNA-蛋白质相互作用(LPI)。同时引入了一种新的特征选择方法(ps:探索性的研究方法会更让审稿人眼前一亮喔!小记者愿意和你一起探讨创新性的研究思路!一起学习创新性的生信思路!),该方法基于FIR网络,可以选择最佳的LPI特征,并进一步提高LPI-DLDN模型的生成能力。
小记者通过这篇文章也开阔了自己的科研思路(ps:来生信日报,和小记者一起打开生信的研究思路吧!),深度学习作为机器学习的一个分支,在生物信息学领域有着广泛的应用。例如通过特征选择,可以去除冗余和无关的特征,提取有用的特征,使得模型具有更好的表达能力和区分度。
















