2022年3月17日,博德研究所Todd R.Golub团队在《Science》(IF2021=47.728)在线发表了题为“Copper induces cell death by targeting lipoylated TCA cycle proteins”的研究论文。
该研究证明铜对于细胞的毒性作用机制不同于已知的其他调节性细胞死亡机制(如铁死亡、细胞焦亡、细胞凋亡等),是一种全新的细胞死亡方式,并将其命名为Cuproptosis——铜死亡。
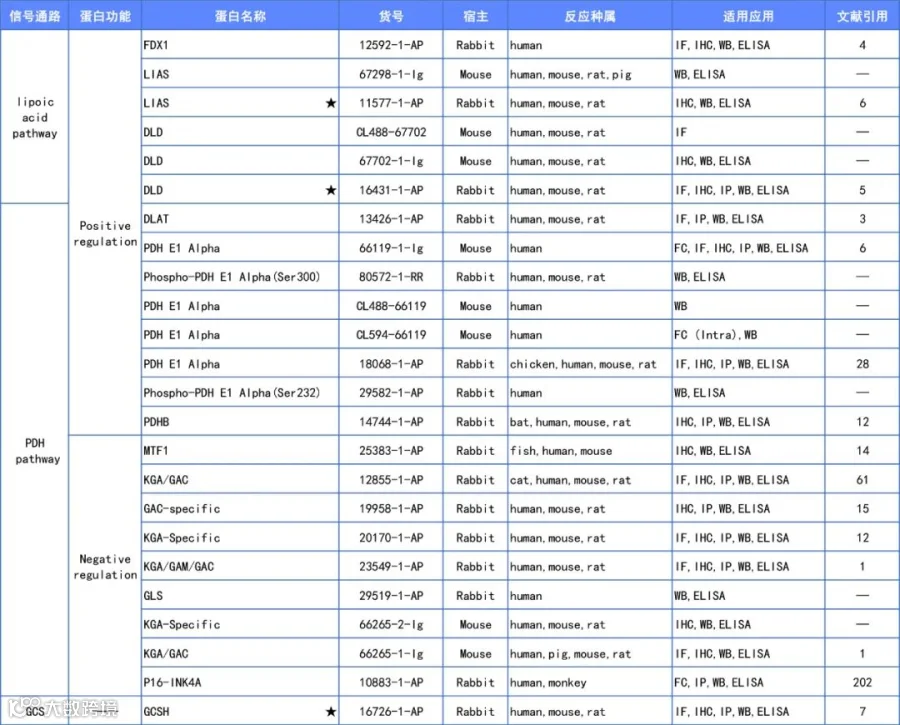
文章中,LIAS、GCSH和DLD三个关键靶标的抗体均源自Proteintech(详情见文末附表)。
铁死亡的热度还居高不下,这又来一个铜死亡。不出意外又是下一个国自然热点。下面小P带大家前排吃瓜学习,了解下文章内容!

原文链接:https://www.science.org/doi/10.1126/science.abf0529
研究背景
1. 作为生命活动必不可少的一种基本元素,铜能够以酶辅基的形式参与到细胞内多种重要的代谢途径中。从细菌到人类细胞,铜都是不可或缺的。
2. 正常情况下,细胞内铜离子的浓度会通过主动稳态机制保持在一个较低水平。
细胞内铜离子浓度过低,会影响许多重要酶的活性及相关代谢过程,影响细胞生存;反之铜离子浓度超过阈值,也会给细胞带来毒害作用,诱导细胞死亡。
Wilson病(hepatolenticular degeneration, HLD;Wilson Disease, WD)和Menkes病(Menkes’disease, MD),分别是由于铜离子代谢相关基因突变,细胞内铜离子过度累积或铜离子浓度过低所导致的。
3. 铜离子载体是一种能与铜结合的小分子,可以将铜运送到细胞中,因此是一种研究铜毒性的有力工具。部分铜离子载体甚至被用于抗癌,杀死癌细胞。
4. 有证据表明,铜离子载体诱导的细胞死亡机制涉及细胞内铜的积累,但目前关于过量铜诱导细胞毒性机制尚不明确。
许多报道之间的结论相互矛盾,或是诱导了细胞凋亡,或是非不依赖半胱天冬酶的细胞死亡,或是诱导活性氧产生,或抑制泛素-蛋白酶体系统,这种跨界效应暗示铜诱导细胞死亡可能靶向的是一种进化上保守的细胞机制。
为了探索这种机制,研究团队对铜诱导细胞毒性这一现象进行了深入的研究。
研究内容
1. 铜诱导一种新形式的细胞死亡
首先,为了明确铜离子载体的细胞毒性是依赖于铜,而不是铜离子载体本身的原因,研究团队分析了多种铜离子载体对细胞的杀伤能力。
结果发现:1)多种结构不同的铜离子结合小分子在数百个细胞系中共享杀伤谱(图1 A)。2)在铜缺乏情况下,细胞对高效铜离子载体Elesclomol具有抗性;补充铜则恢复Elesclomol的细胞杀伤力,补充其他金属元素不具该效果(图1 B)。3)消耗内源铜螯合剂(谷胱甘肽)会增强细胞对Elesclomol的敏感性;补充外源铜螯合剂(四硫代钼酸盐,TTM)则会挽救Elesclomol对细胞的杀伤,其他金属离子螯合剂不具该效果(原文补充数据fig S1)。
表明铜离子载体诱导的细胞死亡主要还是依赖于细胞内铜的积累。
其次,为确认铜离子载体介导的细胞死亡是否受到调节,尤其是在短期暴露后是否会导致不可逆转的持续细胞毒性,研究者继续采用Elesclomol处理细胞。
结果发现:40nM的Elesclomol处理仅2小时,就会导致细胞内铜水平增加15至60倍;且24h后引发细胞死亡(图1 C)。表明铜介导的细胞死亡确实受到了调节,短期暴露后造成了不可逆转的细胞毒性。
接下来,为进一步明确这种受调节的、铜诱导的细胞死亡,是否是已经表征过的细胞死亡方式,研究团队通过关键因子的敲除,以及各种抑制剂处理,一一排除了细胞凋亡、铁死亡、坏死性凋亡和氧化应激等形式的可能性(图1 F&G)。
以上结果表明铜诱导的细胞死亡和已知的细胞死亡途径是不同的机制(图1 H)。
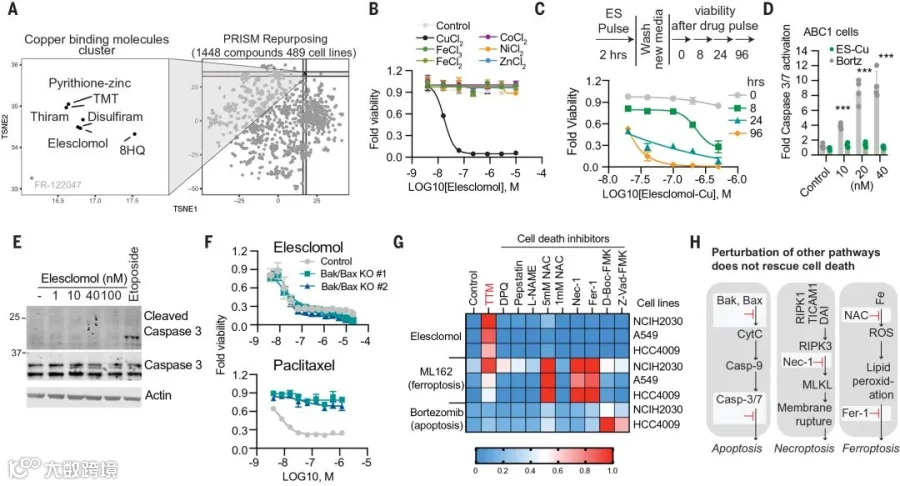
图1 铜诱导的细胞死亡异于其它死亡形式
2. 线粒体呼吸调节铜诱导的细胞死亡
在铜离子载体诱导细胞死亡的实验过程中,团队观察到依赖于线粒体呼吸的细胞,对铜离子载体的敏感性是糖酵解细胞的近1000倍(图2 A)。并且,使用线粒体抗氧化剂、脂肪酸和线粒体功能抑制剂处理,会明显影响细胞对铜离子载体的敏感性(图2 B)。
此外,在电子传递链(ETC)复合物Ⅰ和Ⅱ的抑制剂处理以及线粒体丙酮酸摄取抑制剂处理的条件下,铜离子载体诱导的细胞死亡受抑制,铁死亡不受影响(图2 B)。
更重要的是,线粒体氧化磷酸化解偶联剂FCCP处理对铜离子载体诱导的细胞死亡没有影响。表明该细胞死亡方式是作用于线粒体呼吸,而不是ATP的产生(图2 C)。
随后团队还通过真实的缺氧刺激以及FG-4592激活缺氧诱导因子(HIF)途径作对比,发现只有真实缺氧会降低细胞对铜离子载体的敏感性(图2 D),进一步确认了线粒体呼吸在铜诱导细胞死亡中的关键作用。
随后,进一步分析铜离子载体处理对细胞耗氧率(OCR)的影响,结果显示Elesclomol的处理并不影响细胞的基础耗氧量以及ATP合成耗氧量,仅影响细胞的最大耗氧量,即影响了细胞的最大呼吸能力和备用呼吸能力(图2 E)。暗示铜不直接针对电子传递链(ETC),而可能是针对三羧酸循环(TCA)。
为验证上述观点,团队对Elesclomol处理过的细胞进行代谢物分析,结果Elesclomol敏感的细胞中,许多TCA循环相关代谢物的代谢物失调呈时间依赖性增加,但在Elesclomol抵抗的细胞中则没有这个现象。
以上结果建立了铜离子载体诱导的细胞死亡和线粒体代谢之间的紧密联系(图2 F)。
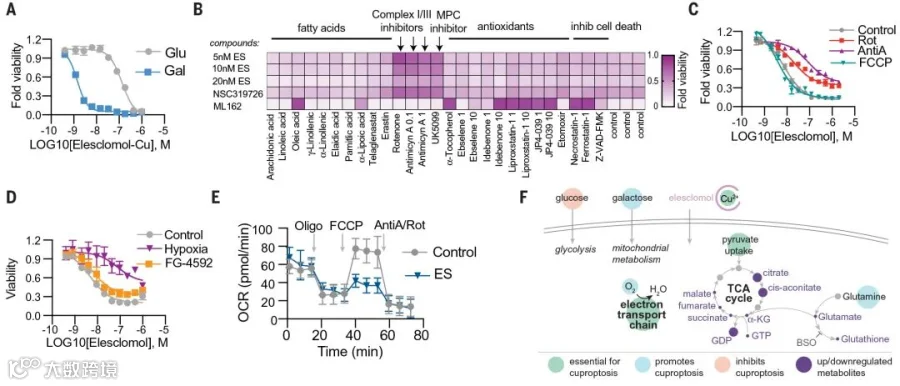
图2 线粒体呼吸调节铜诱导的细胞死亡
注:通过依次加入Oligomycin(ATP合酶抑制剂)、FCCP(氧化磷酸化解耦剂)、 Antimycin A和Rotenone(电子传递抑制剂)测定OCR,Oligomycin加入前的耗氧量代表细胞的基础耗氧量;Oligomycin加入后ATP合成途径受阻,减少的耗氧量代表用于ATP合成的耗氧量;FCCP加入后的耗氧量代表细胞的最大耗氧量,它与基础耗氧量之间的差值代表细胞还具有的呼吸潜力,即备用呼吸能力;Antimycin A和Rotenone的加入则完全阻断了线粒体呼吸,代表非线粒体呼吸的耗氧量。
3. FDX1和蛋白硫辛酰化是铜诱导细胞死亡的关键介质
为明确铜诱导细胞死亡的具体代谢路径,团队进行了全基因组CRISPR-Cas9功能缺失筛选,并采用不同结构的铜离子载体进行处理,以提高筛选的普遍性(图3 A-C)。
结果筛选到7个基因,它们的敲除可以明显挽救两类铜离子载体介导的细胞杀伤(图3)。包括FDX1(一种还原酶、能将Cu2+还原成Cu+,且是Elesclomol的直接靶标),LIPT1、LIAS、DLD(硫辛酸途径的三个关键酶),DLAT、PDHA1、PDHB(丙酮酸脱氢酶复合体的三个组分,该复合体是硫辛酰化蛋白靶点)。
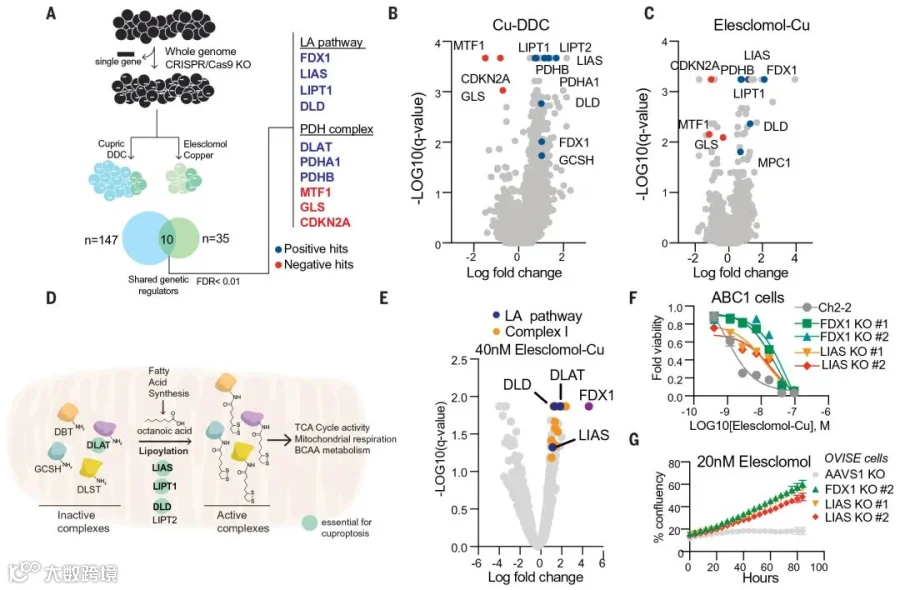
图3 FDX1和蛋白硫辛酰化是铜诱导细胞死亡的关键介质
4. FDX1是蛋白硫辛酰化的上游调控因子
蛋白质硫辛酰化修饰(Protein Lipoylation)是一种从细菌到哺乳动物均保守的蛋白质翻译后修饰。α-硫辛酸含有双硫五元环结构,通过酰胺键修饰底物蛋白的赖氨酸残基从而形成硫辛酰化蛋白。
迄今为止,哺乳动物中已知的4个硫辛酰化修饰蛋白DBT(支链α-酮酸脱氢酶复合体组分)、GCSH(甘氨酸裂解系统蛋白H)、DLST(α-酮戊二酸脱氢酶复合体组分)、DLAT(丙酮酸脱氢酶复合体组分),都是细胞中重要代谢酶复合物的核心组分,调节碳进入三羧酸循环(图3 D)。
团队发现,不管是敲除FDX1还是硫辛酰化相关酶均可以保护细胞免遭铜毒害,这让他们怀疑FDX1可能是蛋白质硫辛酰化的上游调节因子。
为验证以上猜想,团队首先在癌症依赖性图谱(Cancer Dependency Map,网址:www.depmap.org)中找到了二者高度关联的证据(图4 A);随后他们对208例人类肿瘤标本进行FDX1和硫辛酸免疫组化染色,结果显示FDX1和硫辛酰化蛋白的表达高度相关(图4 B-C);第三,他们还使用硫辛酸特异性抗体确认了FDX1的敲除会导致DLAT和DLST的蛋白硫辛酰化缺失(图4 D),还会导致细胞呼吸水平下降,与LIAS敲除结果一致(图4 E)。
并且,通过代谢物分析发现,FDX1的缺失会导致丙酮酸和α-酮戊二酸的积累,琥珀酸的消耗(图4 F)。这与预期结果一致,表明TCA循环在丙酮酸脱氢酶复合体(PDH)和a-酮戊二酸脱氢酶复合体(a-KDH)两个位置受到抑制,暗示两个酶复合体的硫辛酰化修饰受阻。
同时在硫辛酰化途径中,还观察到LIAS的关键底物S-腺苷甲硫氨酸(SAM)的积累(图4 F),进一步说明FDX1在上游参与调节蛋白的硫辛酰化过程。
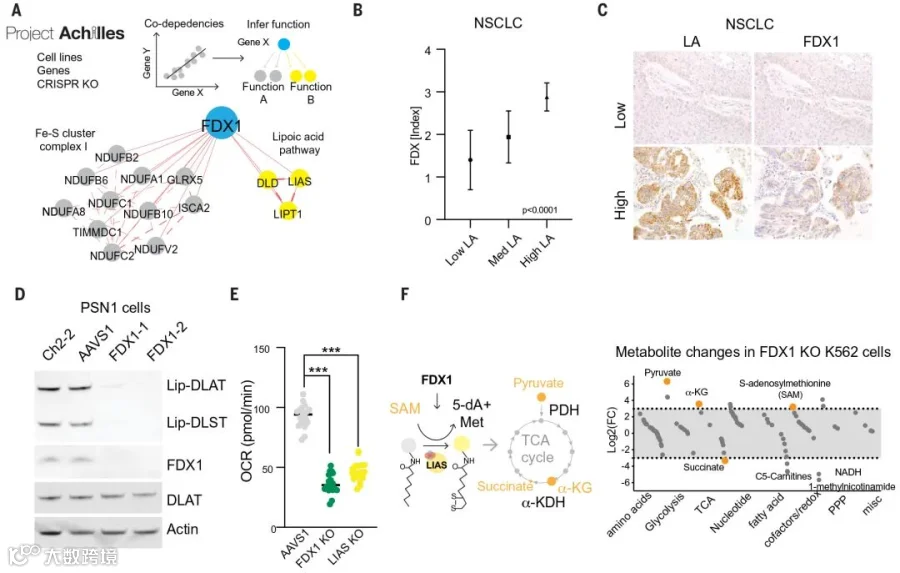
图4 FDX1是蛋白质硫辛酰化的上游调节因子
5. 铜直接结合硫辛酰化 DLAT 并诱导其寡聚化
以上实验结果充分证实了线粒体代谢关键酶的硫辛酰化修饰与铜诱导细胞死亡的紧密联系,但是并没有找出二者之间的直接联系。
为此团队猜想铜离子可能会与硫辛酰化的蛋白直接结合。通过有利硫辛酸与铜的结合试验,发现二者解离常数为10-17,暗示上述猜想是有可能的。
为进一步验证该猜想,团队采用FDX1敲除细胞和正常细胞的裂解液纯化DLAT和DLST两个蛋白,让他们与偶联铜(或镍、钴)的树脂结合,结果发现,正常细胞中的DLAT和DLST(硫辛酰化状态),会直接与铜离子结合,FDX1敲除细胞中的DLAT和DLST(去硫辛酰化状态),不与铜结合(图5 A-B)。
充分表明蛋白的硫辛酰化修饰,是直接结合铜的必要条件。
团队还注意到,铜与硫辛酰化蛋白的结合,会导致蛋白的寡聚化,Elesclomol的处理会增加DLAT寡聚体的水平(图5 B-C)。
通过免疫荧光发现,Elesclomol处理对DLAT的聚集有着显著的促进作用,并且这种聚集在硫辛酰化缺陷的FDX1敲除细胞中减少(图5 F-H)。
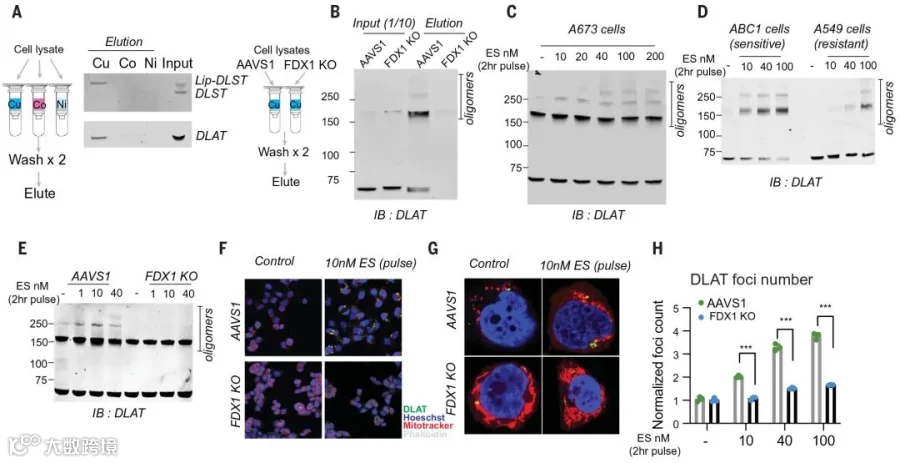
图5 铜直接结合硫辛酰化 DLAT 并诱导其寡聚化
此外质谱分析还发现,铜离子载体处理还会导致Fe-S簇蛋白的水平降低,并且这个过程依赖于FDX1蛋白的存在;还有HSP70等蛋白水平的上升,表明蛋白毒性应激的发生(图6-1 A-C)。其中Fe-S簇蛋白水平的变化与在细菌和酵母中观察到一致,均表明铜会使Fe-S簇蛋白不稳定。
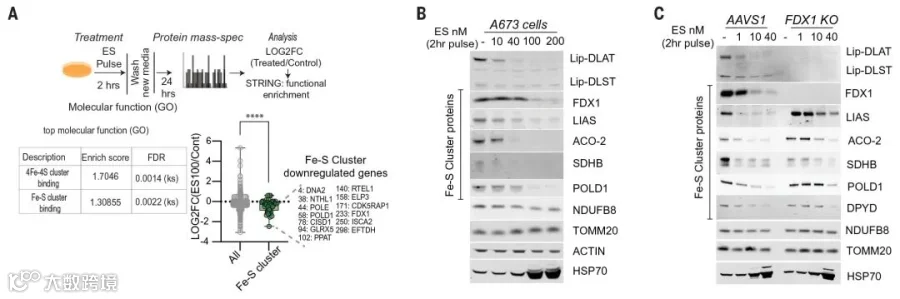
图6-1 铜诱导Fe-S簇蛋白的不稳定和蛋白毒性应激
至此铜离子载体诱导的细胞死亡机制基本梳理清楚,即铜离子载体导致细胞内铜离子水平超过阈值,过量铜促进脂酰化蛋白的聚集和Fe-S簇蛋白的不稳定,导致蛋白毒性应激并最终导致细胞死亡。
6. 铜诱导细胞死亡机制与铜稳态失调的遗传模型相同
文章开头我们提到正常细胞内,存在一个铜水平的主动稳态机制,这个机制主要依赖三个铜转运蛋白SLC31A1(CTR1)、ATP7A和ATP7B,SLC31A1负责铜摄入,ATP7A和ATP7B负责铜转出。他们分别是铜失调综合征Wilson’s disease和Menke’s disease中发生突变的基因编码产物(图6-2 D)。
为探索铜离子载体诱导的细胞杀伤与这些自然发生的铜稳态障碍是否是同一种分子机制导致,团队在HEK 293T和ABC1细胞中过表达了SLC31A1,这显著增强了细胞对铜的敏感性(图6-2 E),与野生型细胞被铜离子载体处理后的结果一致:蛋白硫辛酰化减少,Fe-S簇蛋白水平降低,HSP70水平升高(图6-2 F)。
更重要的是,在过表达SLC31A1的细胞中,铁死亡、坏死性凋亡和凋亡抑制剂并不能阻止铜诱导的细胞死亡(图6-2 G);铜螯合剂、FDX1敲除和LIAS敲除各自部分拯救了铜诱导的细胞死亡(图6-2 G-H);并且LIAS的敲除还能缓解内源性铜螯合剂谷胱甘肽耗尽诱导的细胞死亡(图6-2 J)。
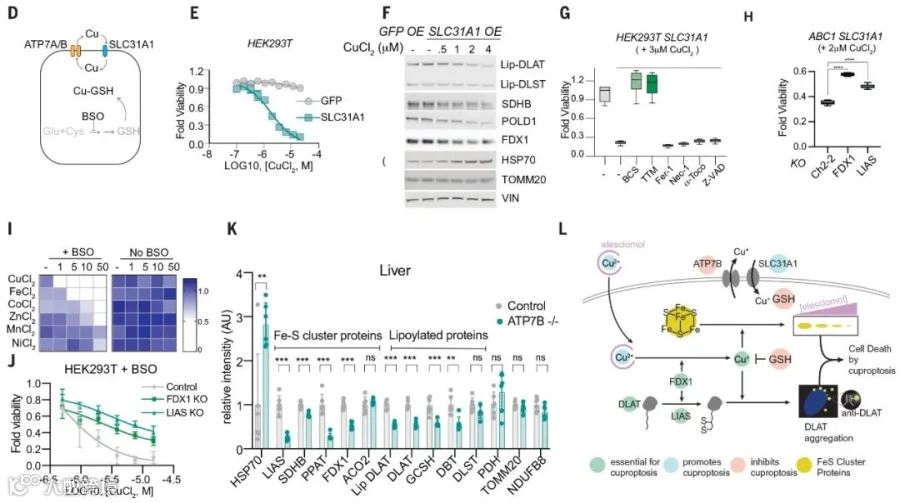
最后,采用Wilson’s disease小鼠模型发现,与野生型小鼠相比,老年Atp7b缺陷小鼠的肝脏中蛋白的硫辛酰化水平和Fe-S簇蛋白的含量均有显著降低,Hsp70蛋白则有明显增加(图6-2 K)。
以上多种模型均显示,铜过量导致的细胞死亡机制和铜离子载体诱导的细胞死亡机制是同一种机制。
为此,研究团队建议将这种全新的细胞死亡机制命名为铜死亡——Cuproptosis。
了解了铜死亡机制后,团队还对铜离子载体治疗肿瘤的临床数据进行了分析,发现正在进行的Elesclomol的3期联合临床试验总体结果并不理想,但其中一部分血浆乳酸脱氢酶(LDH)水平低的患者显示了抗肿瘤活性的证据。低LDH反映了细胞对线粒体代谢的更高依赖性(与糖酵解相反),这与团队揭示的铜死亡机制一致。
同时团队还观察到FDX1和硫辛酰化蛋白的丰度在多种人类肿瘤中高度相关,并且具有高水平硫辛酰化蛋白的细胞系对铜诱导的细胞死亡敏感。
文章小结
总而言之,该研究揭示了铜死亡是一种全新的、受调控的细胞死亡方式,铜离子通过直接结合三羧酸循环途径中的硫辛酰化组分,导致硫辛酰化蛋白的异常聚集,Fe-S簇蛋白的丢失,从而引起蛋白毒性应激反应并导致细胞死亡。
抗体推荐
铁死亡热度还在持续攀升当中,这又出来一个铜死亡。想必接下来的几年,我们会看到大量的铜死亡相关文章出现。
值得一提的是,上述研究中引用了Proteintech的LIAS、GCSH和DLD三个抗体(列表中星号标记),感谢研究团队的信任!
小P也根据咱们的抗体库将文中筛选到的靶标做了汇总,具体详情见下表,希望咱们的相关产品也能在铜死亡研究领域中给大家带来帮助!