

表观遗传机制通过动态调控染色质结构和DNA可及性,在基因表达调控中发挥核心作用。以ChIP-seq、CUT&Tag、CUT&RUN和ATAC-seq为代表的二代测序技术,通过捕获蛋白质-DNA互作位点和染色质开放区域,已成为解析表观遗传调控网络的关键工具。
然而尽管这些技术在研究中取得了显著成果,但是它们在实际应用中仍面临诸多挑战。首先样本间差异是一个普遍存在的问题,例如不同样本的细胞数量、染色质状态或实验处理条件可能导致结果的不一致性;其次实验偏差难以避免,这里的偏差包括了抗体的特异性、实验操作的标准化程度以及测序文库构建的效率等。因此,在这些实验中标准化和定量问题就尤为突出了,特别是在比较不同样本或不同实验条件下的数据时,如何实现准确的定量分析成为难题。
为了解决这一问题,研究人员提出了一种叫做加标Spike-in的方法。Spike-in最早是在RNA-seq技术中引入并广泛应用的。它的出现主要是为了解决RNA-seq数据分析中的标准化和定量问题。随着其成功应用,Spike-in逐渐扩展到其他高通量测序技术中,成为提高数据可靠性和可比性的重要工具。Spike-in可作为实验内参,用于后续的数据归一化,从而能够准确解释样本之间每个基因组区域的信号是增加还是减少。
通过使用Spike-in control,研究人员可以校正样本产量和测序深度的变化,确保观察到的信号差异反映的是真实的生物学变化,而非技术性伪影。这种方法显著提高了基因组学和转录组学研究中比较分析的可靠性和准确性。
在ChIP-seq、RNA-seq等高通量测序实验中,Spike-in校正是确保数据准确性和可比性的关键步骤,加标Spike-in主要有以下三个原因:
1. 建库过程需要通过PCR扩增
在进行需要高通量测序的组学实验时,我们通常需要将提取到的 DNA/RNA 构建成测序文库(library),这个过程包括 PCR 扩增,而这一步能引入一些非线性偏差,改变原始样本中各片段的相对丰度(取决于片段长度、GC含量、结构等等)。例如原本在WT组中某个peak区域富集得特别强,但在PCR扩增时因为GC含量高扩增效果差,那它在最终文库中就可能被低估了。
2. 每个样本的文库通常会用相同的量测序
测序平台在上机之前,会尽量将每个样本的文库量调成一致,这样有助于获得类似数量的 reads。然而这个做法忽略了样本本身的真实生物差异。
3. 标准的生物信息学方法是基于标签计数进行归一化
在数据分析中,常见的归一化方法(如RPM, Reads Per Million)假设每个样本的总测序标签数(tag counts)是相同的,并将数据标准化到相同的尺度。然而,这种归一化方法只有在基因组上信号增加的总和等于信号减少的总和时才有效,而这种情况在真实的生物学实验中很少发生。例如,某些基因或基因组区域的信号可能显著增加,而其他区域的信号变化较小,导致总信号发生变化。如果使用传统的归一化方法,可能会错误地解释数据。通过添加Spike-in,可以根据Spike-in的信号比例调整样本间的标签计数,从而更准确地反映样本间的真实差异。
在2014年的一篇cell reports上,David等人提出了一种叫ChIP-Rx(ChIP with reference exogenous genome)的方法来定量哺乳动物细胞的动态表观基因组图谱(PMID: 25437568)。他们选择了进化距离适中的黑腹果蝇(Drosophila melanogaster)基因组作为哺乳动物细胞的外参,因为果蝇基因组经过充分研究并且注释完善,同时与人类或小鼠基因组的同源性小,可避免测序读长错误比对,同时组蛋白修饰保守性高。
其原理如图1所示,图1 A显示了标准的ChIP-seq工作流程,chromatin state A和B分别是具有全部(Full)和一半(Half)的组蛋白修饰的人类表观基因组(红色圆点表示组蛋白修饰,蓝色圆点表示核小体)。进行 ChIP实验、测序并使用标准归一化方法(RPM)分析作图,可得ChIP-seq的峰(蓝色)。将峰占总读数的百分比进行比较,发现差异不大。
图1B是具有Spike-in的ChIP-seq数据工作流程。chromatin state A和B和图1A一致,但是引入了固定量的参考表观基因组(即Spike-in chromatin,红色圆点表示组蛋白修饰,橙色圆点表示参考物种的核小体)。在ChIP实验、测序并将ChIP序列读数标准化为样品中参考基因组读数的百分比(reference-adjusted RPM,RRPM)来进行分析。将得到ChIP-seq信号峰进行比较,发现峰之间存在50%的差异。这种方法称为具有参考外源基因组的ChIP(ChIP-Rx)。
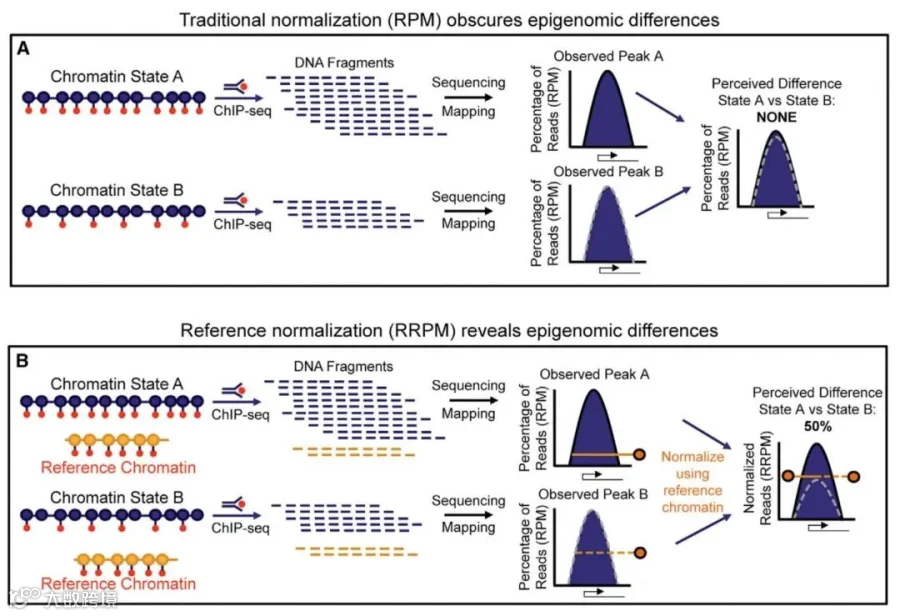
图 1. ChIP-Rx加标Spike-in原理示意图
ChIP-Rx方法是在体系里添加来自不同物种(黑腹果蝇)的恒定数量的参考细胞,从而对不同生物样品中的组蛋白修饰进行全基因组定量比较。沉淀的参考DNA与沉淀的实验DNA一起测序,将参考序列读数作为内参,使得其可以在ChIP实验中作为一种标准化的方法。该方法最终取决于实验ChIP抗体在参比和实验物种中识别目标组蛋白修饰的能力。
相比于传统的ChIP-Rx方法,Active Motif开发了一种新方案来进行样本间的Spike-in control。与ChIP-Rx方法相似的是,我们在实验体系中加标了黑腹果蝇的染色质;然而与ChIP-Rx方法不同的是,该方案创新性地引入果蝇特异性H2Av抗体,与实验抗体形成正交检测系统(实验抗体仅识别目标物种修饰如人H3K27me3,H2Av抗体特异性结合果蝇染色质,二者互不干扰)。
该方案中,Spike-in反应的建立是通过将目标染色质(例如人类或其他物种的染色质)、目标抗体、少量果蝇染色质以及果蝇H2Av特异性抗体混合在一起完成的。果蝇Spike-in染色质以等量添加到每个反应中,而H2Av抗体的作用是下拉(pull down)每个反应中的少量果蝇染色质。测序后,测序标签(tags)会被分别比对到目标染色质对应的基因组以及果蝇基因组上。对于每个样本,统计唯一比对到果蝇基因组的标签总数,并利用这些数据生成校正因子(例如DMSO标签数/抑制剂标签数)。然后,使用这些校正因子对目标染色质的标签计数进行归一化处理。
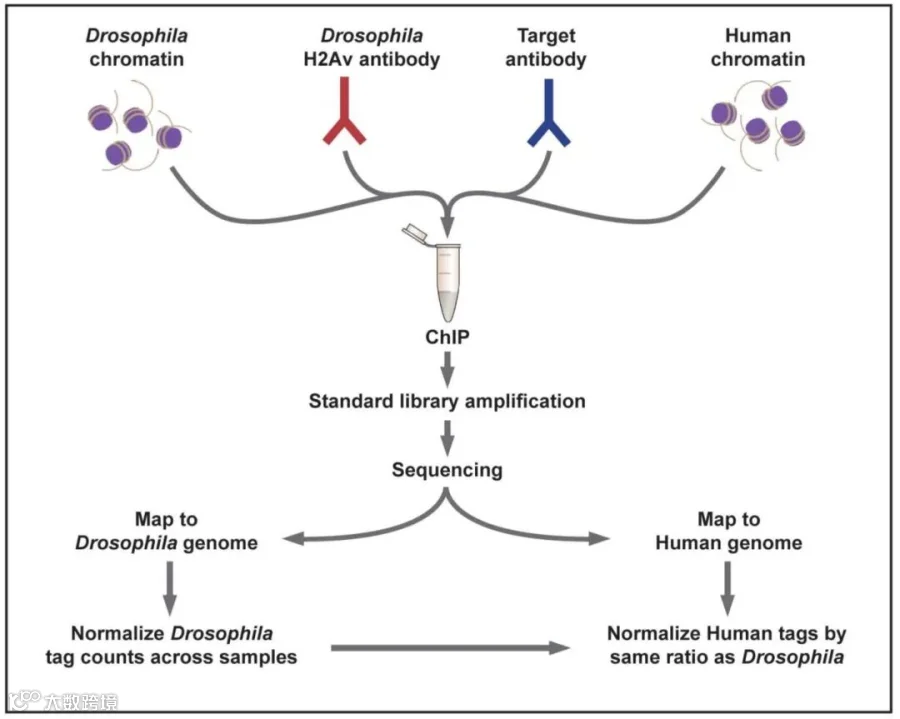
图2:Active Motif ChIP-seq加标Spike-in方案示意图
Active Motif也使用EZH2抑制剂模型验证了该方案。EZH2是H3K27最主要的特异性组蛋白甲基转移酶(HMT),在转录抑制中起重要作用。多个研究都已确定EZH2是诸如前列腺、乳腺癌和血液系统恶性肿瘤等许多人类恶性肿瘤的候选肿瘤靶点;在多种肿瘤中均发现EZH2体细胞突变,该突变改变了EZH2的底物特异性并增加了全局H3K27me3水平;目前许多EZH2的小分子抑制剂被视为一种有前途的癌症疗法,正在临床上开发。因此为了更好地了解人类癌细胞中受 EZH2 抑制影响的分子事件,监测整个基因组中H3K27me3模式的抑制剂诱导改变非常重要。然而,由于H3K27me3是一种全局表达的组蛋白修饰,通过标准ChIP-seq方案和数据分析方法检测EZH2抑制剂诱导的H3K27me3水平全局差异是困难的。
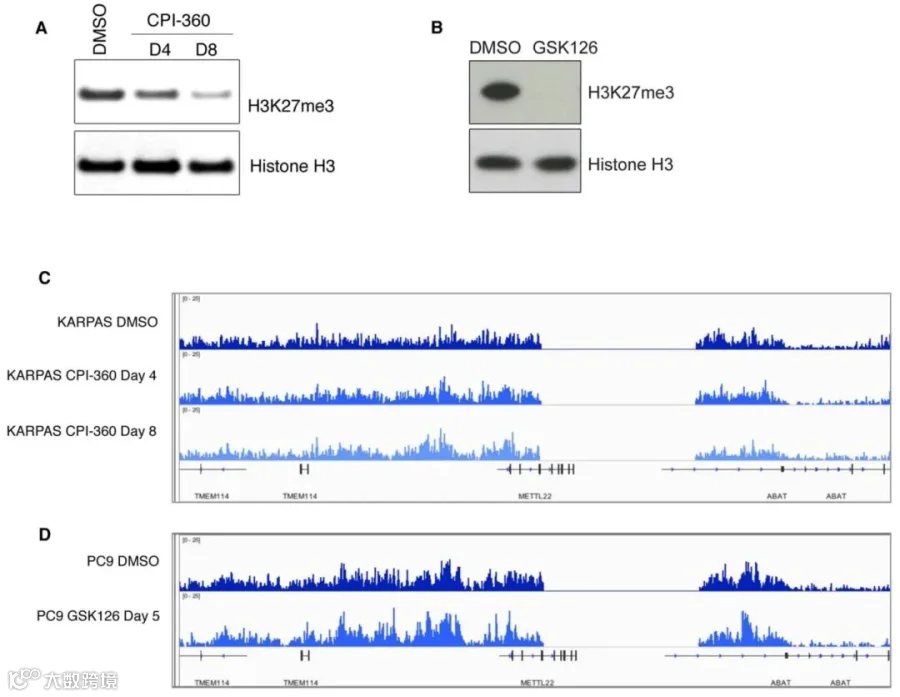
图3:EZH2抑制剂介导的H3K27me3水平降低实验
(A)Western blot显示用 1.5 μM CPI-360 处理4天和8天的KARPAS-422细胞中整体H3K27me3水平降低。
(B) Western blot显示用1 μM GSK126处理5天的PC9细胞中整体 H3K27me3 水平降低。
(C、D)使用IGV显示 H3K27me3 ChIP-seq结果。与载体处理的对照相比,在CPI-360(C)和GSK126(D)处理的KARPAS-422和PC9细胞中未检测到明显差异。
如图3所示,为了更好地了解EZH2抑制剂诱导的H3K27me3水平全局差异的检测难点。我们使用两种EZH2 抑制剂(CPI-360、GSK126)分别处理了两种细胞(KARPAS-422、PC9),和DMSO处理相比,WB实验显示两种细胞中H3K27me3加药后的表达均降低(图3A,B),然而标准ChIP-seq实验显示,DMSO组和药物处理组未见显著差异(图3C,D)。为了验证ChIP实验是否成功,分别对H3K27me3 ChIP实验建库前后的DNA进行ChIP-qPCR检测。可以发现建库前的DNA在EZH2抑制剂处理后,相比于DMSO组,MYT1启动子区域的H3K27me3富集减少(图4A,B)。而使用建库后DNA的ChIP-qPCR实验表明,DMSO和EZH2抑制剂处理的样品之间的差异不存在(图4C,D)。
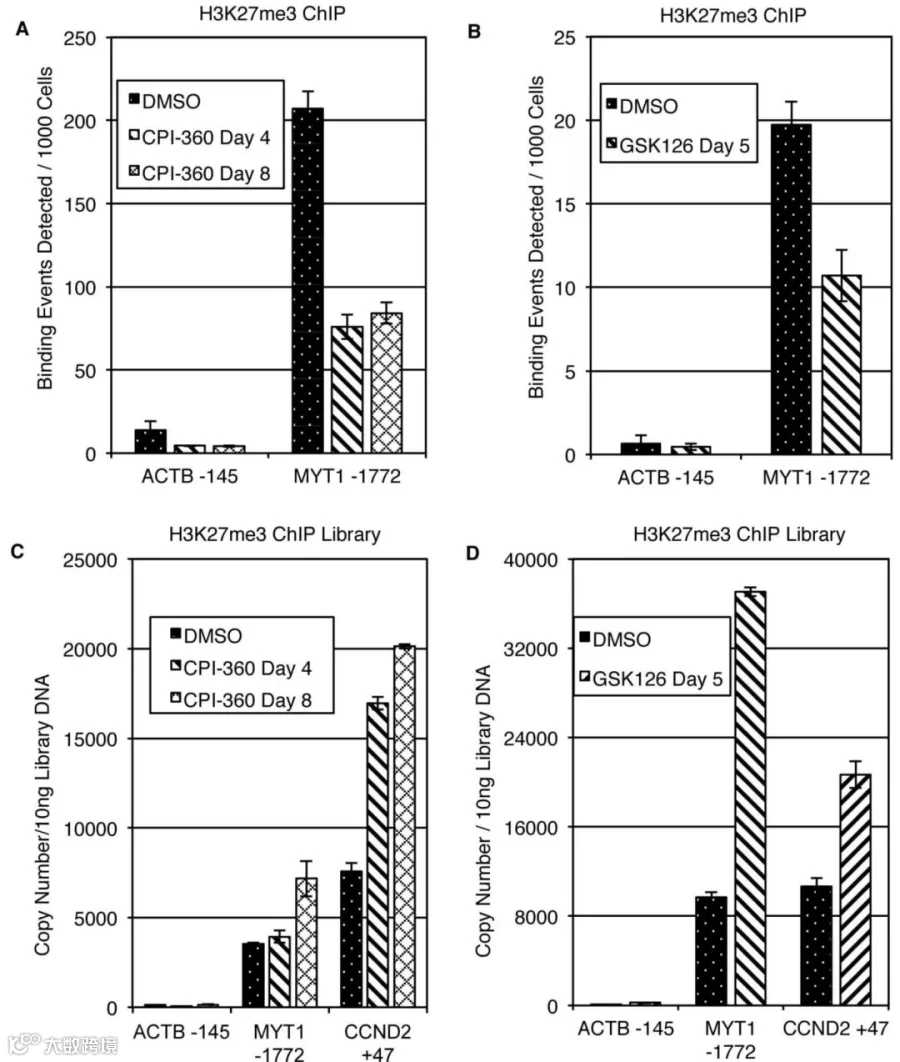
图4:使用H3K27me3 ChIP实验建库前后的DNA进行ChIP-qPCR实验
(A)使用经EZH2抑制剂CPI-360处理的KARPAS-422细胞的染色质进行ChIP- qPCR检测。使用阳性对照引物MYT1的qPCR显示,在抑制剂存在下,H3K27me3的结合率降低。
(B)使用经EZH2抑制剂GSK126处理的PC9细胞的染色质进行ChIP- qPCR检测。使用阳性对照引物MYT1的qPCR显示,在抑制剂存在下,H3K27me3的结合率降低。
(C)使用15个PCR扩增循环从(A)生成文库。稀释文库DNA并使用 MYT1和CCND2的阳性对照引物进行qPCR检测。
(D)如(C)所述从(B)生成文库,并使用MYT1和CCND2的阳性对照引物进行qPCR检测。所有实验均表示为两个独立实验的平均值,qPCR一式三份,x̄±SD。ACTB启动子作为所有实验的阴性对照。
这些结果表明,除了随后的生物信息学分析外,标准文库生成方案对于准确定量表示来自不同处理条件的生物样品中的全局组蛋白PTM变化可能不是最佳选择。因此我们使用加标Spike-in策略来重复上述实验。如图5所示,加标了Spike-in后的H3K27me3 ChIP-seq数据可以正确反映出EZH2抑制剂对H3K27me3下游结合染色质的影响。CPI-360处理4天和8天导致KARPAS-422细胞中整个基因组的H3K27me3水平显着降低(图5A),而用GSK126处理5天后在PC9细胞中观察到的情况也类似(图5B)。这种ChIP-seq加标Spike-in方案可以成为ChIP-seq标准化的广泛适用解决方案。采用这种ChIP-seq Spike-in方案或其他类似策略将揭示更多以前未检测到的染色质状态和染色质因子结合率的差异,从而增加ChIP-seq作为生物学研究工具的力量。
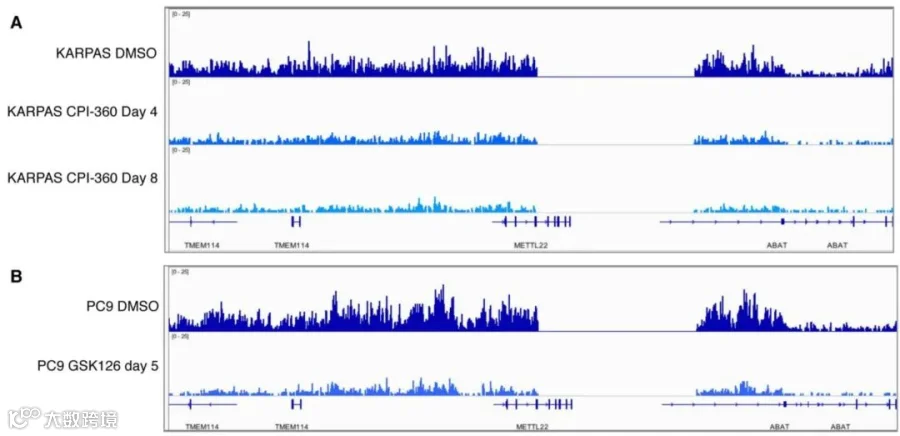
图5:加标Spike-in后的H3K27me3 ChIP-seq数据。
(A)用CPI-360处理4天和8天后,加标Spike-in的H3K27me3 ChIP-seq IGV图像。
(B)用GSK126处理5天后,加标Spike-in的H3K27me3 ChIP-seq IGV图像。
CUT&Tag和CUT&RUN这两个技术和ChIP-seq类似,它们都是用于蛋白-DNA互作的表观遗传学工具,也需要引入Spike-in来帮助研究人员进行定量和标准化。根据Active Motif ChIP的Spike-in策略,我们也开发了CUT&Tag和CUR&RUN的Spike-in策略。和ChIP Spike-in策略不同的是,这里将黑腹果蝇的染色质(Spike-in chromatin)改为了黑腹果蝇的细胞核(Spike-in Nuclei),而识别组蛋白变体H2Av的抗体不变。这样,Spike-in Nuclei就可以掺入实验组的细胞中,并在实验组进行Target-Specific抗体孵育时,添加Spike-in抗体(图6)。这样就可以和实验组一起进行整个CUT&Tag或CUT&RUN实验,方便后续生物信息学分析。
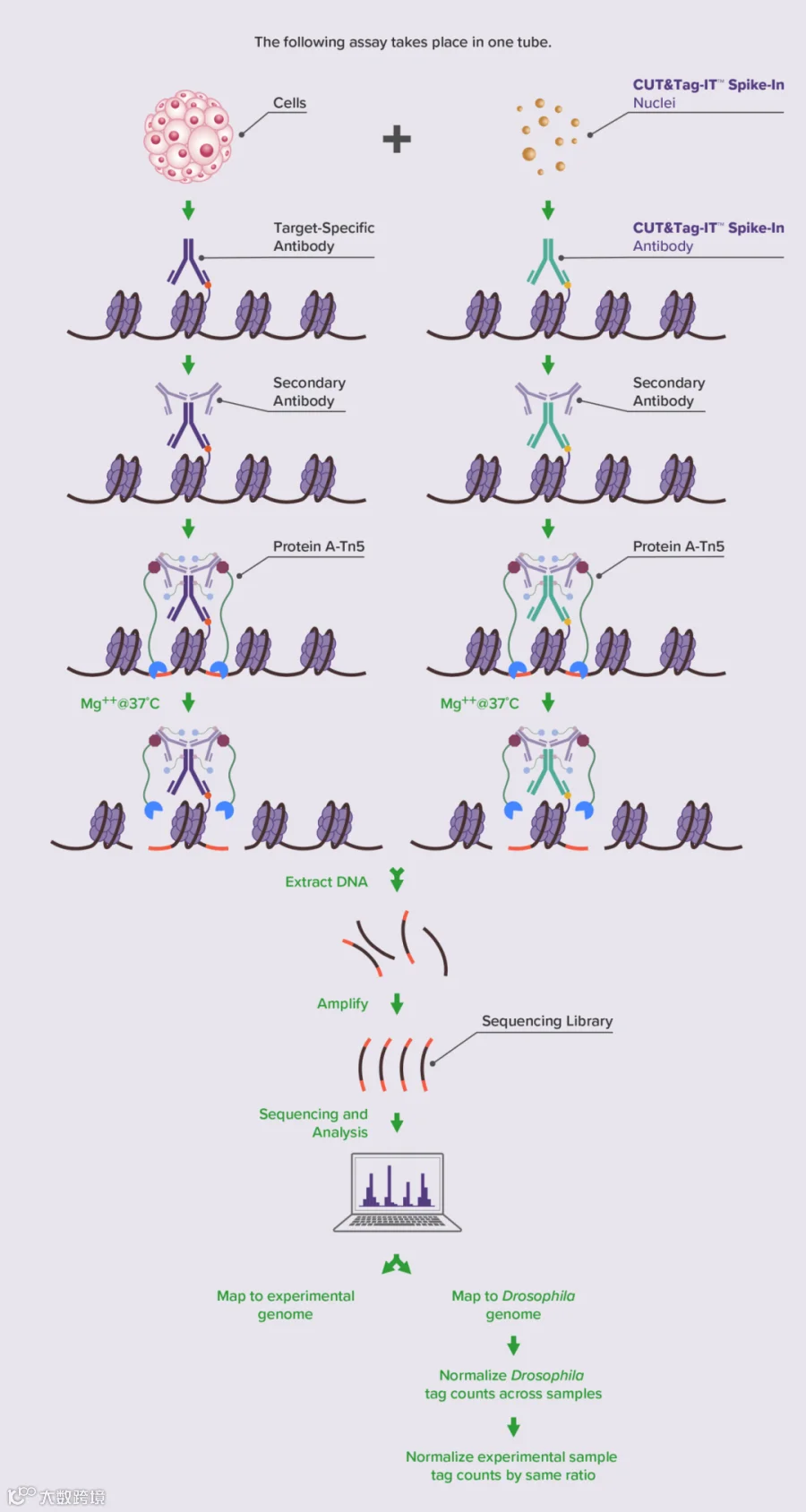
图6:CUT-Tag Spike-in策略
对于ATAC-seq技术,由于其实验本身不需要使用Target-Specific抗体,所以我们提供了黑腹果蝇的细胞核(Spike-in Nuclei),与CUT&Tag策略类似,可以将Spike-in Nuclei掺入实验组细胞,一起进行整个ATAC实验。Spike-in Nuclei将随实验组一起被Tn5切割,PCR扩增和上机测序。测序结果中的果蝇来源的染色质将成为样本比较和归一化的参考。
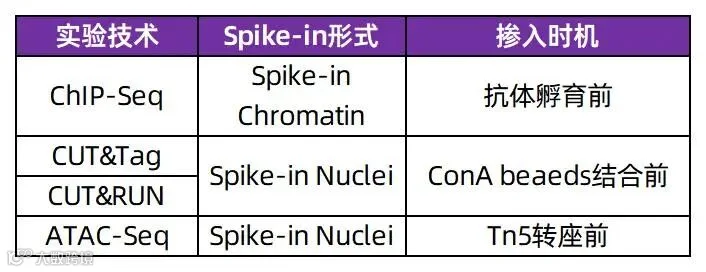
表1. Active Motif针对不同技术开发Spike-in策略
在加标Spike-in时,我们应该注意以下5点:
1) 确保所有样品中均应等量的加标Spike-in。
2) 在实验早期添加Spike-in,以便控制在整个实验和文库生成过程中的实验偏差。
3) 通过预实验确定Spike-in读长占比(约2-5%),避免添加过多Spike-in,以免实验组的文库受到影响,导致最终测序数据由来自Spike-in的标签主导。
4) 加标的Spike-in在最终数据中应是唯一可识别的。
5) 加标的Spike-in可以普遍应用于ChIP / CUT&Tag / CUT&RUN / ATAC反应,与实验样品染色质的性质无关,也独立于实验特异性抗体。
Active Motif是一家全球领先的生命技术公司,专注于表观遗传学领域20余年,致力于为生命科学研究和药物开发提供创新的试剂和整体解决方案。Active Motif提供ChIP/CUT&Tag/CUT&RUN/ATAC一系列实验的Spike-in策略作为表观遗传药物开发工具。希望我们的Spike-in校正策略可以帮助研究者深入理解更多生物学问题,开发更多药物。
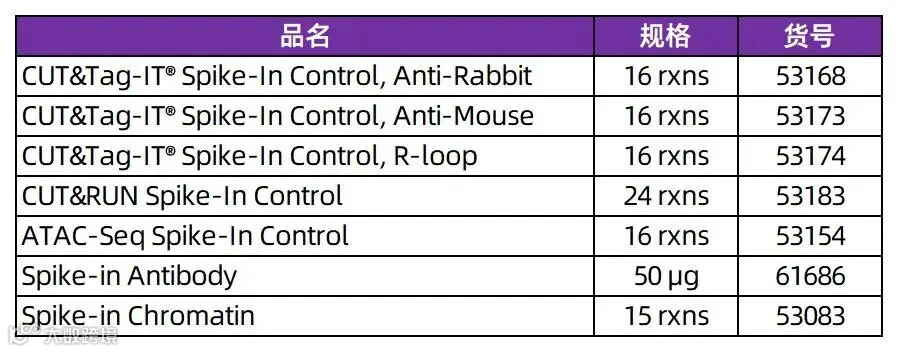
目前,下列Spike-in现货产品可享7折特惠,助力表观研究。各位小伙伴如果对Active Motif的产品感兴趣,或者想了解更多的表观产品,都可以添加文末小助手咨询,或直接与Proteintech当地销售渠道沟通!
Active Motif Spike-in Normalization 产品


扫码添加云南总代理云南泽浩客服微信






往期文章推荐
