

之前小编整理了tagSNP的筛选教程分享给大家,如果有没看到的盆友们可以在本文最下方寻找链接查看~
今天给大家带来的是针对筛选过程中遇到的问题/errors整理的一些FAQ/Trouble Shooting:
Q1
千人计划查找vcf文件时,输入基因名后显示找到了多个Gene Product,该选哪一个?
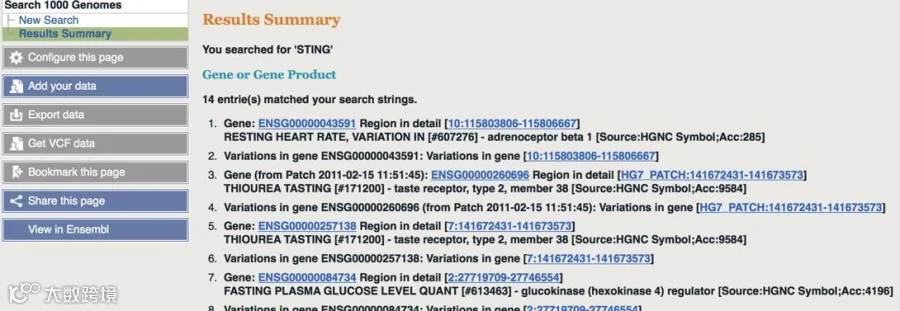
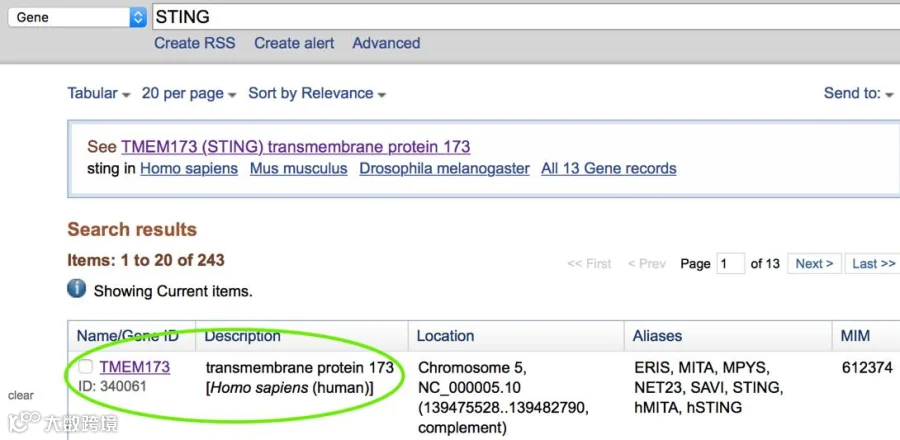
可在NCBI数据库上查找目的基因的Gene Symbol,看Human物种,如下图,STING基因的Gene Symbol名为“TMEM173”,然后再回到千人网站去找对应结果。
Q2
直接在NCBI数据库中查找基因所在染色体区域可以吗?
可以的,但一定注意必须选择GRCh37版本的,这样才能与1000 Genomics数据对应。
Q3
安装haploview时提示安装失败,提示java文件损坏该怎么办?
可参考https://wenku.baidu.com/view/4f246e7ac5da50e2524d7fd3.html链接中教程。Q4
Q4
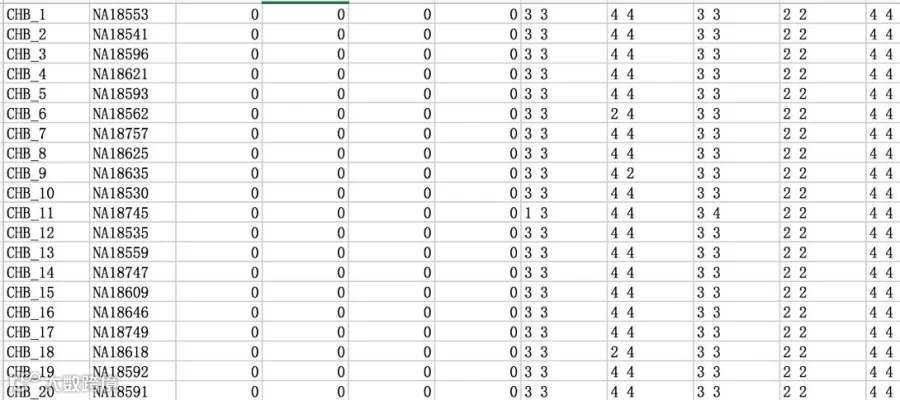
数据导入Haploview软件时报错,提示“Column number mismatch in ped file. line 101”,怎么办?

表示ped文件中存在数据缺失,解决方案:ped文件是可以通过txt文档打开的,打开后复制所有数据至excel表格中(比txt方便查看和编辑),即可看到下图所示数据,“line 101”指的就是“CHB_101”排数据存在缺失,下拉表格,再往后找,把缺失的位置填上“0 0”(注意中间有空格,表示该位点对应的该样本分型结果不明),保存,再导出后缀为.ped的txt文档即可。
注:由于不能识别A/C/G/T,故用数字代替,1代表碱基A,2代表碱基C,3代表碱基G,4代表碱基T。
Q5
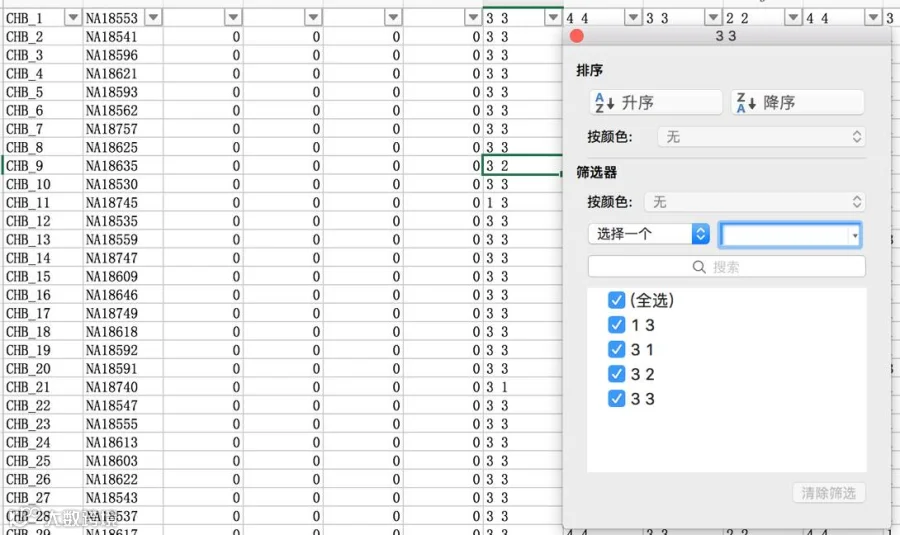
数据导入Haploview软件时报错,提示“More than two alleles at marker 1”,怎么办?

表示marker 1存在不止两个等位,因为Haploview软件只可分析二等位结果,解决方案;同上述操作相同,将ped文件复制到excel表格中,找到Marker 1 column(从第7列开始算),比对该列数据,可通过筛查的方法找到哪个数据出了问题,找到后可将数字改成“0 0”。
Q6
如果想把5’UTR和3’UTR区域也包含进去找TaqSNP,怎么办?
在千人/NCBI上找到的染色体区域基础上,前后各扩大2kb即可(500bp-2kb都可以,没有明确的标准)。
Q7
为什么同一基因、相同染色体区域、软件参数相同条件下查找出来的tagSNP不一样?
因为Haploview在选tagSNP的时候是随机的,每次结果不一样,但对结果是没有影响的,比如说苹果、橙子、香蕉都代表水果,那么随机挑其中两样得到的结果也是水果,这种的话就会有3种组合,不过一般最终倾向于选择有功能的SNP往下研究。
以上即为小编自己在操作过程中遇到的问题,参照上述基本可顺利完成tagSNP筛选这一步骤,最后祝有这方面课题的小伙伴们科研顺利~
往期分享:
1. 干货:如何利用1000 Genomes数据挑选tagSNP?
