

2019年8月12日Nature Biotechnology杂志发表了PacBio与谷歌、NIST等多家知名研究机构和公司的合作文章“Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome”,这篇文章最早发布于今年1月的预印本文献bioRxiv上,我们当时也发布过一篇公众号文章来进行介绍“高准确率的长读长测序 | 使用PacBio 13.5kb CCS文库组装高精度人类基因组结果发布”。
PacBio的单分子实时(SMRT)测序技术经过多年的发展,已经使酶读长度增加到平均100 kb。通过优化和平衡酶读长与插入片段的长度,我们开发了一种基于CCS(Circular Consensus Sequencing)的高准确率长读长测序模式——HiFi Reads。能够在提供长读长reads(10-20kb)的同时兼顾准确性(Q30,99.9%准确率)。HiFi Reads的生成原理请参考下面动图。

PacBio HiFi Reads的生成原理
为了评估这些长而且准的HiFi Reads的性能,就需要对一个标准基因组进行测序。我们选择了Genome in a Bottle的人类参考样品HG002/NA24385,该样品之前已经使用多种测序技术进行过测序,已经建立了非常准确的基准变异数据库,在定义的基准区域内HG002基因组的预估准确度可以达到每400万碱基对只有一个错误。我们对HG002基因组DNA文库进行了28倍深度的HiFi Reads测序,检测了PacBio HiFi Reads识别基因变异和进行基因组从头组装的能力。
HiFi Reads 对于SNVs和Indel的检测能力
实验结果显示,当使用HiFi Reads进行单碱基突变(SNVs)和短的插入缺失变异(Indel, <50bp)检测,并使用Google的DeepVariant分析流程时,PacBio对SNVs和Indel的检测F1分数分别可以达到99.91%和95.99%。

HiFi Reads 对于结构变异的检测能力
在结构变异(>50bp的插入或缺失)检测方面,一直是PacBio SMRT测序的强项。果然HiFi Reads 的表现也没有让人失望。对结构变异检测的敏感性可以达到96.13%,特异性可以达到95.99%。此外,与其它长读长测序技术相比,PacBio HiFi Reads可以精确的确定结构变异的断点和长度。
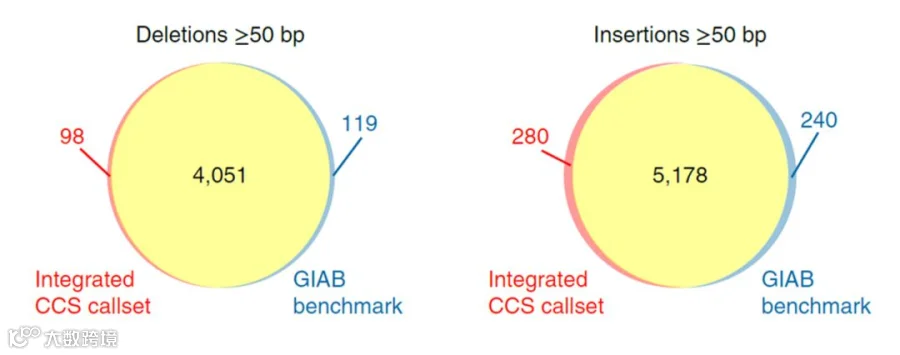
PacBio HiFi Reads检测结构变异结果与GIAB benchmark的对比,黄色区域是二者共有变异,红色是HiFi Reads独有的突变,蓝色是GIAB benchmark独有的变异。可以明显的看出二者吻合度非常高。
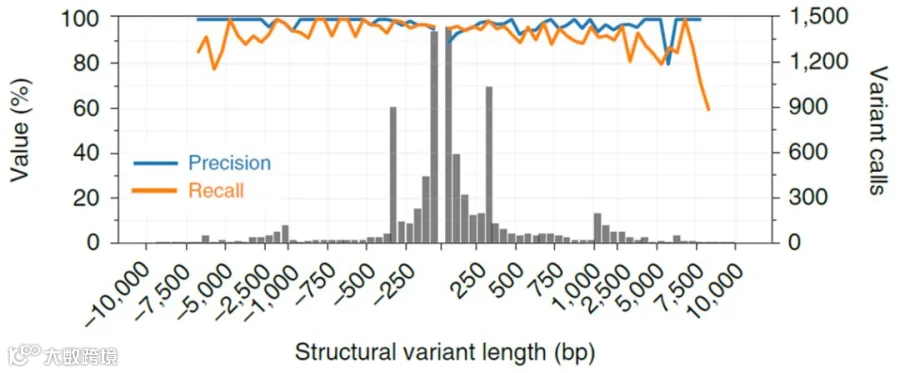
按照结构变异不同长度的敏感性和特异性分布,X轴正值表示插入片段长度,负值表示缺失片段长度。蓝色线代表检测敏感性,橙色线代表检测特异性
使用HiFi Reads 进行基因组De Novo组装的能力
在基因组从头组装方面,使用HiFi Reads应用FALCON、Canu和wtdbg2算法分别对HG002基因组进行了从头组装,结果显示组装质量均较高,contig N50超过15Mb,并且与HG002标准结果高度一致,吻合率达到99.9983%(Q47.7)。这一组装结果无论是对比单独Nanopore测序组装方案(Q24.6)或Nanopore测序+Illumina测序组装方案(Q28.8)都具有明显优势。

ONT=Oxford Nanopore, CLR=PacBio continuous long reads
从下图可以看出,使用PacBio HiFi reads组装出的contig质量达到Q50的有60%左右。而单独使用Nanopore reads组装的HG001基因组 contig 质量达到Q50 的是0% (没有任何contig 能达到Q50)。即使使用Nanopore + Illumina 纠错后混装方案达到 Q50 的contig 也只有15%。
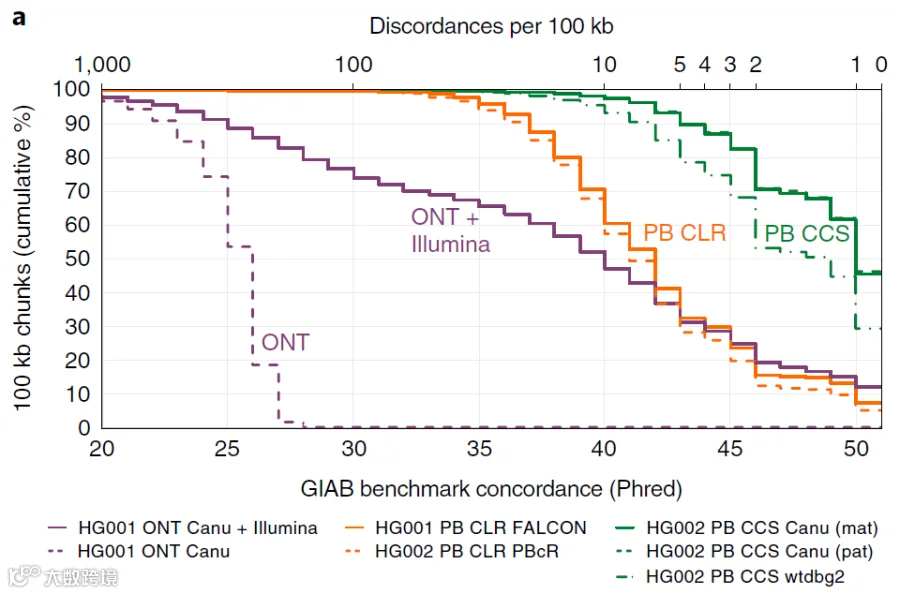
Contig chunk 在质量分数上的累积分布曲线,PB=PacBio, ONT=Oxford Nanopore, CLR=PacBio continuous long reads
几乎所有(99.6%)的变异都能够通过PacBio HiFi测序进行单倍型分型。能够进行准确单倍型分型的前提是测序reads必须同时具备长读长和高准确性。PacBio HiFi reads的原始平均准确度可以达到99.8%,已经不需要NGS的数据进行打磨纠错就可以进行高质量的基因组组装和分辨单倍型信息。
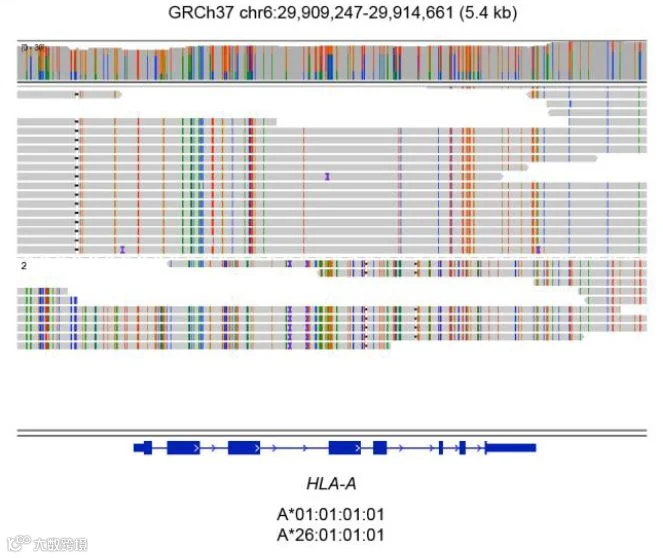
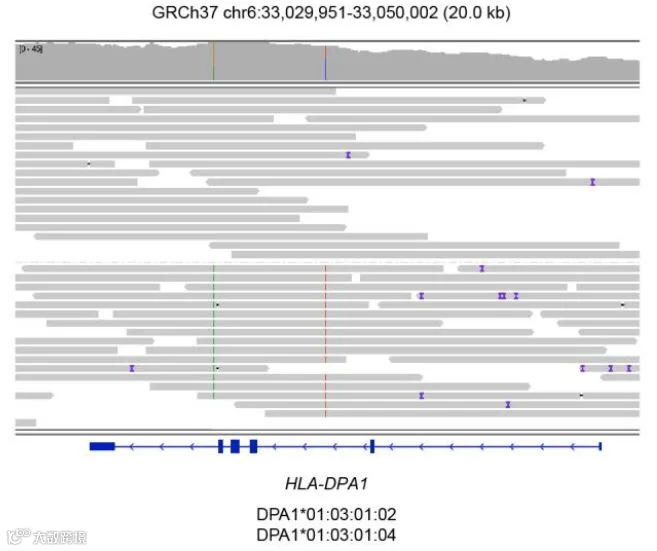
HiFi reads提供HLA I类和II类基因的分型和全长分辨率(可向右滑动查看)
由于PacBio SMRT测序没有GC偏好性,将HiFi Reads测序结果比对到人GRCh37基因组发现能够覆盖到很多通过短读长测序无法覆盖的区域,其中就包括152个与医学相关的基因。
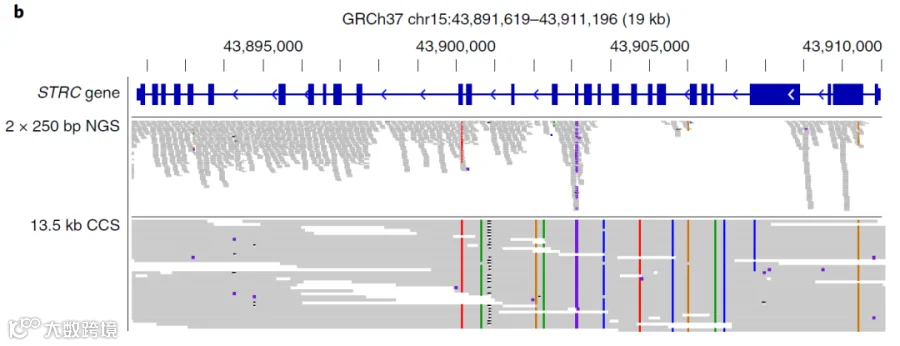
STRC基因使用短读长测序reads与PacBio HiFi reads的比对结果,可以看到PacBio SMRT测序能够更好的覆盖这些难测区域
根据研究人员估计,通过HiFi reads对HG002的测序,可以发现GIAB benchmark中存在的2,434个测序错误。部分测序错误经过人工检查确认。


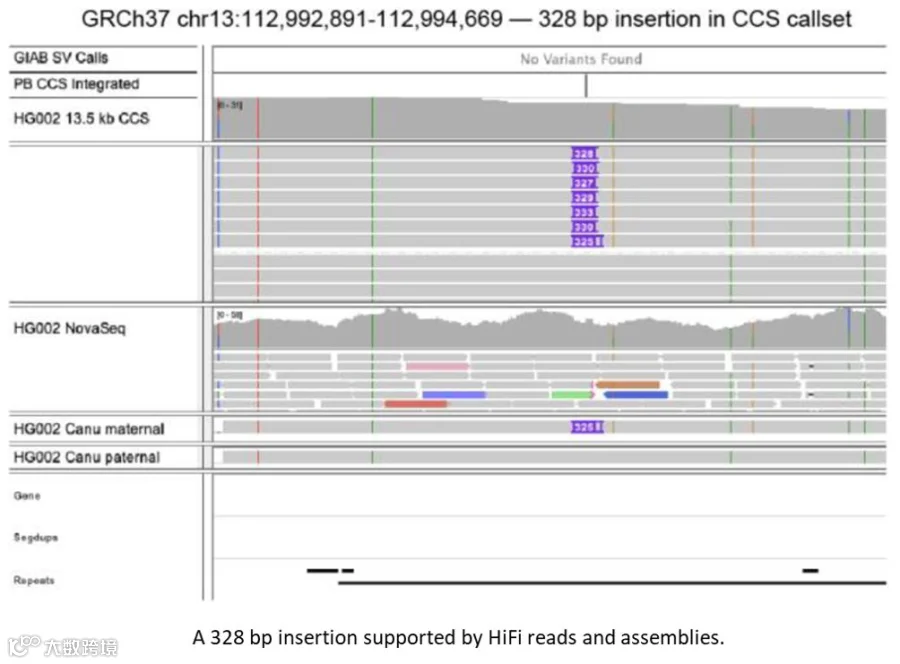
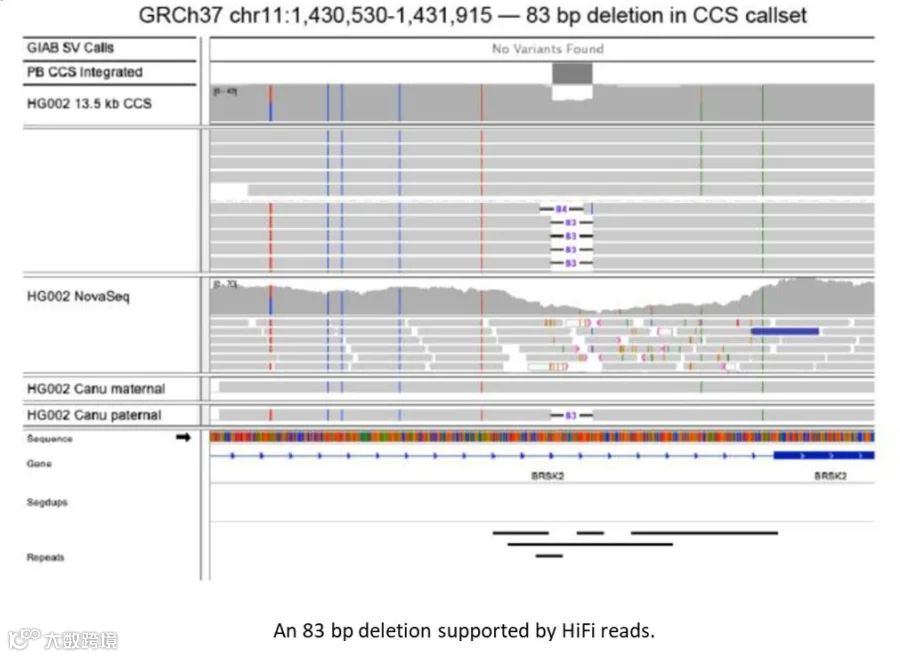
HiFi reads发现的GIAB benchmark中可能存在的错误(可向右滑动查看)
PacBio HiFi reads带来了一种全新的测序数据模式,可以在保持长读长的同时提供与NGS相同的准确性。通过这项研究,充分验证了HiFi reads在基因组从头组装,SNVs和Indel检测以及结构变异检测中的表现。使得只使用一种测序平台就能够检测基因组上所有的变异类型成为可能,这将大大拓展PacBio SMRT测序的应用领域以及改进现有的测序结果,相信也会为各类基因组研究带来更多新的发现。
PacBio SMRT测序原理
