
经典名句“有机不成环,起码错一半”告诉我们:任何反应都需要分析其“成环”的可能性,尤其对初学者而言。
本文深入分析其原因。
一、“成环”案例
“成环”又分为两种:
1. 形成环状中间体或过渡态
① 在Smiles重排中,芳环经分子内SN2 Ar发生重排,反应经环状过渡态,如图1;
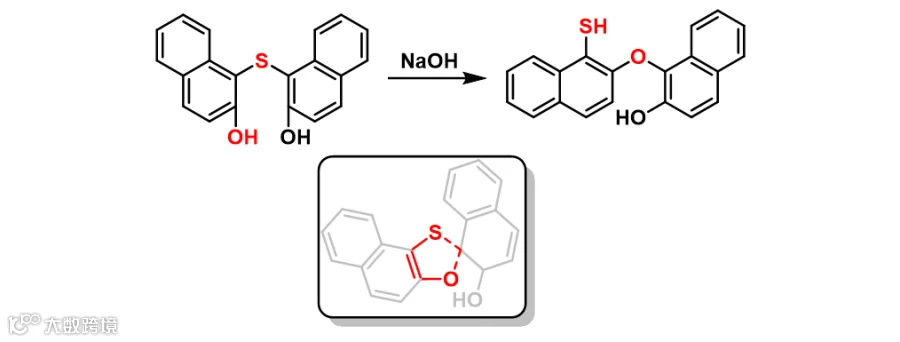
图1:经五元环过渡态的Smiles重排反应
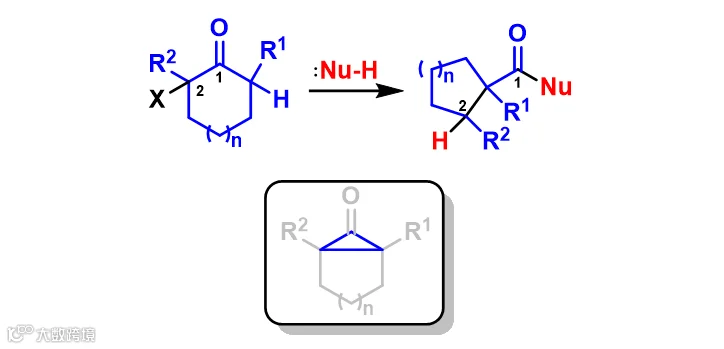
② 在Favorskii重排中,经三元环中间体得到缩环产物(图2);
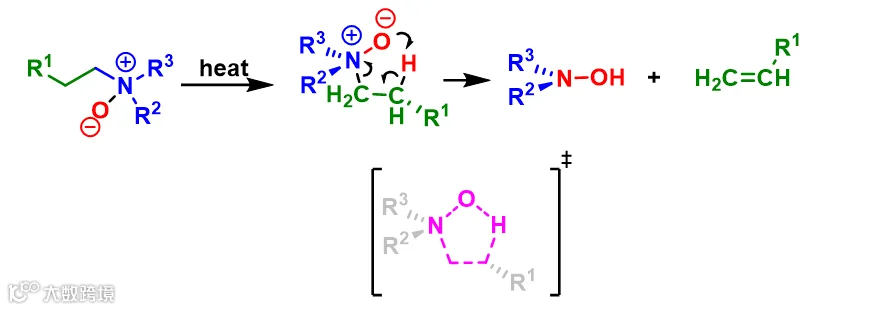
③ 在Cope消除中,氮氧化物在加热下经五元环过渡态最终得到羟胺和烯烃(图3)。
2. 形成环状产物
① 卤代醇经分子内亲核取代关环生成环醚(图4)。

③Bischler-Napieralski 异喹啉合成法(图6)、Combes 喹啉合成法(图7)等通过分子内反应构建芳环环系。
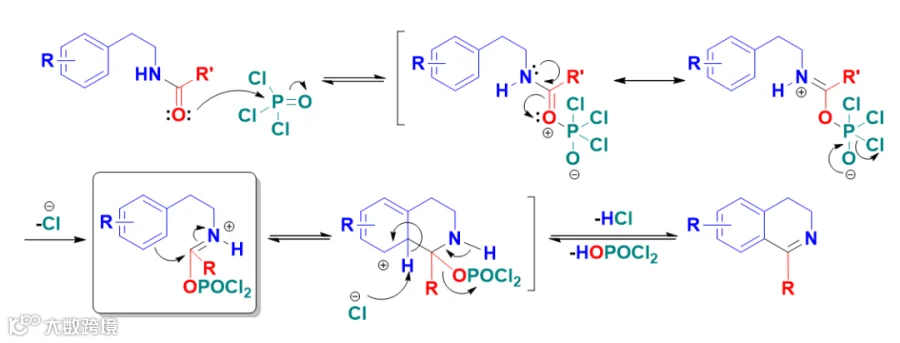
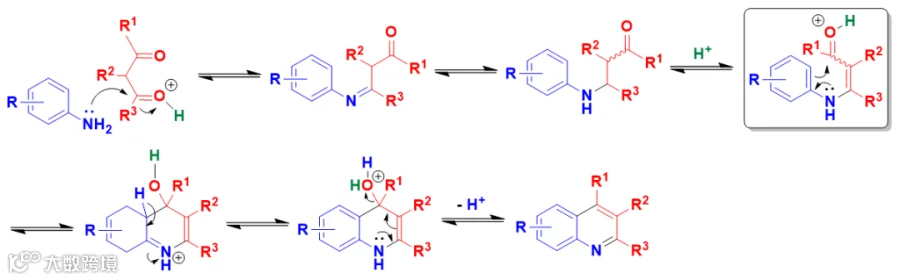
图7:Combes 喹啉合成法
二、“成环”驱动力
之所以存在众多“成环”案例,是因为该过程本身具有非常高的驱动力:大量“成环”过程都是分子内反应(参考图1~图7),而分子内反应“天然”具有高活性,尤其跟分子间反应对比。
1. 碰撞原理
反应可视为原子或分子彼此间的碰撞,对于分子内反应,碰撞位点之间的距离比分子间反应更近,发生碰撞的“机会”更大,进而使反应发生的“机会”更大。如图8:分子间反应和分子内反应的碰撞模型对比。
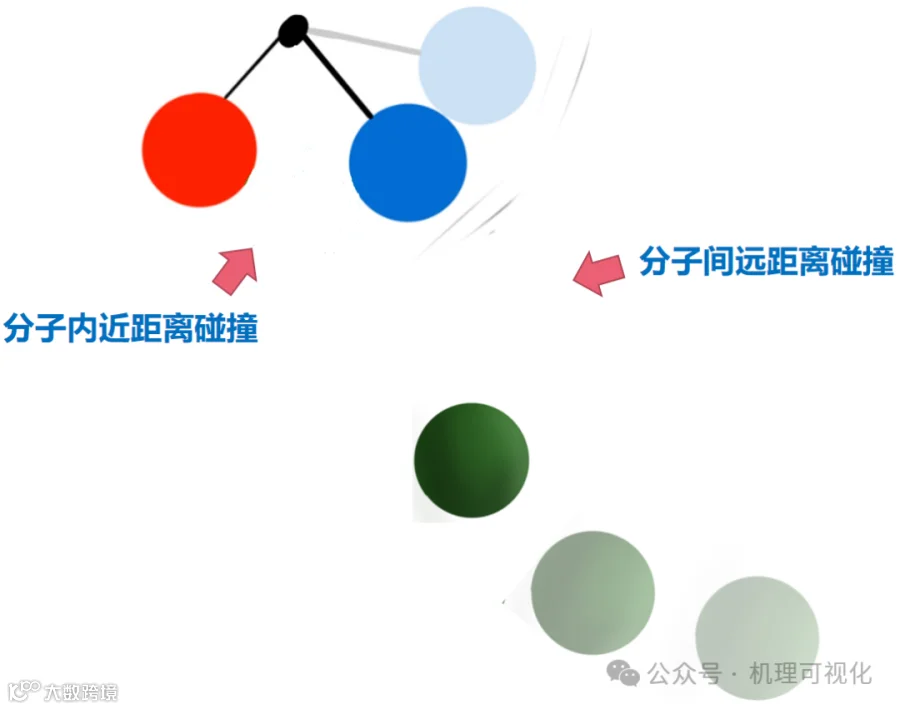
图8:分子间和分子内反应的碰撞模型对比
2. 熵效应
举图9为例,卤代醇环化制备环醚是典型的分子内反应,其仅由一分子底物参与反应,却生成两个产物分子:环醚和氢溴酸。

图9:环醚的制备
产物分子数量增加意味着体系能量受到更多粒子分散,即熵增,可视为图10:自由奔跑,即系统混乱度增加。

图10:自由奔跑(系统混乱度增加,熵增)
而该反应可能存在的分子间SN2竞争反应则要慢得多,因为它要以熵降低的代价生成过渡态,见图11:SN2过渡态两分子合一,熵不利。
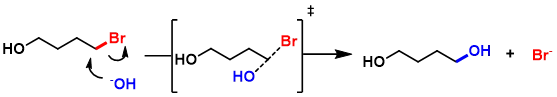
图11:分子间副反应熵不利
对标图10,熵不利可视为图12:被卡住,即系统混乱度降低。

图12:被卡住(混乱度降低,熵不利)
所以对于上述底物,相比分子间取代,通过分子内反应形成环醚一般会更快,其他分子内反应同理。
上述原理为“成环”提供了大量驱动力。
三、“成环”特点
正因为分子内反应活性通常高于相应的分子间反应,所以很多分子间难发生的反应,只要引入“成环”过程,使之成为分子内反应,就容易发生了:
1. 常规的分子间SN2芳香亲核取代需要芳环足够缺电子,但Smiles重排作为经环状过渡态的分子内反应,却甚至部分富电子芳环都能反应(图13);
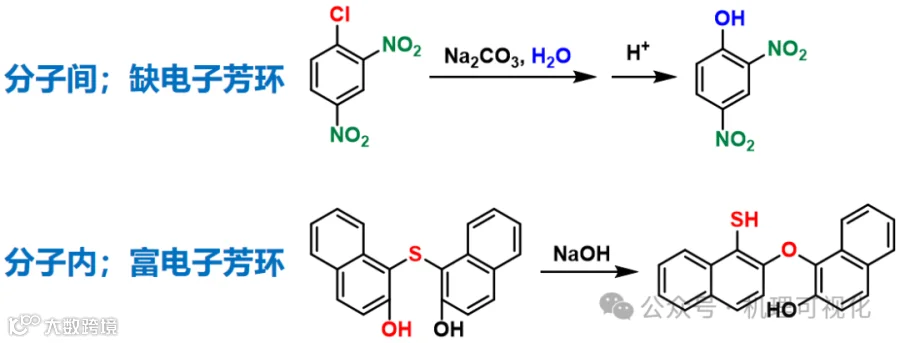
图13:分子间和分子内SN2芳香亲核取代
2. 分子间Williamson合成法通常无法在NaOH 作碱条件下得到产物,因NaOH 碱性不足以拔掉醇氢,但分子内反应在NaOH 下却可顺利构建三元环系(图14);
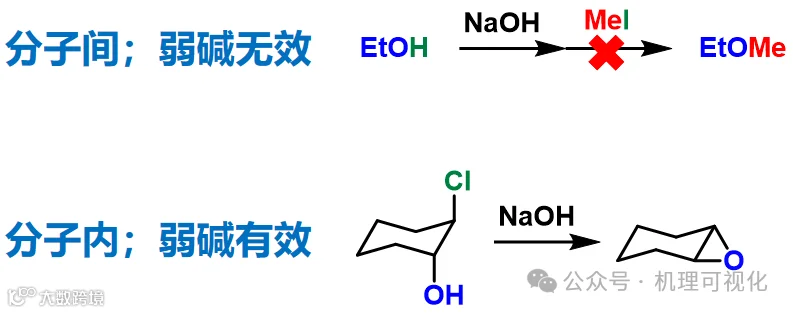
图14:分子间和分子内Williamson合成法
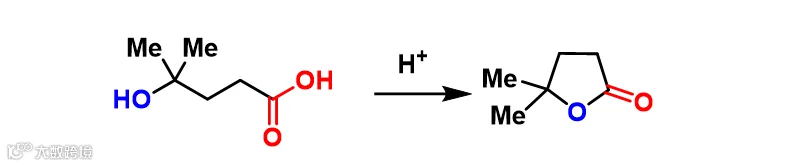
3. 分子内内酯化环化反应的转化率通常高于常规的分子间酯化反应(图15)。
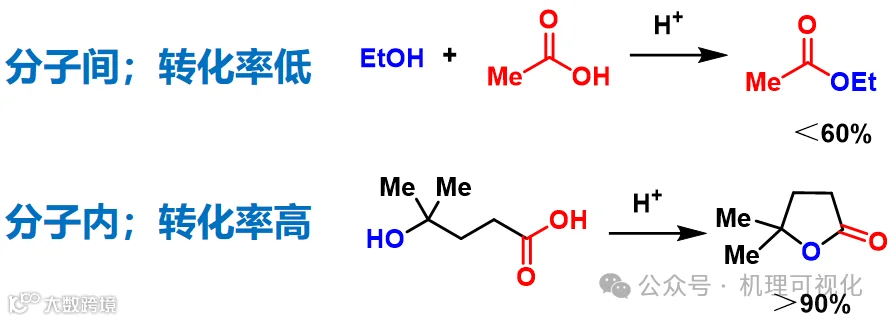
图15:分子间和分子内酯化
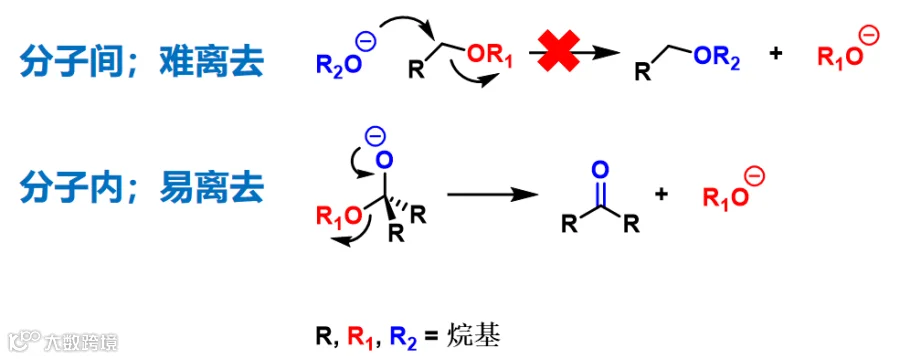
分子内反应天然具有的高活性推动了“成环”的发生,由此,分子内反应也成为构建环系的重要方法。
注意
1. 成环亦可经分子间反应如环加成,后文讨论;
2. 因熵效应及环张力等原因,9~11元环相对难通过分子内反应合成。