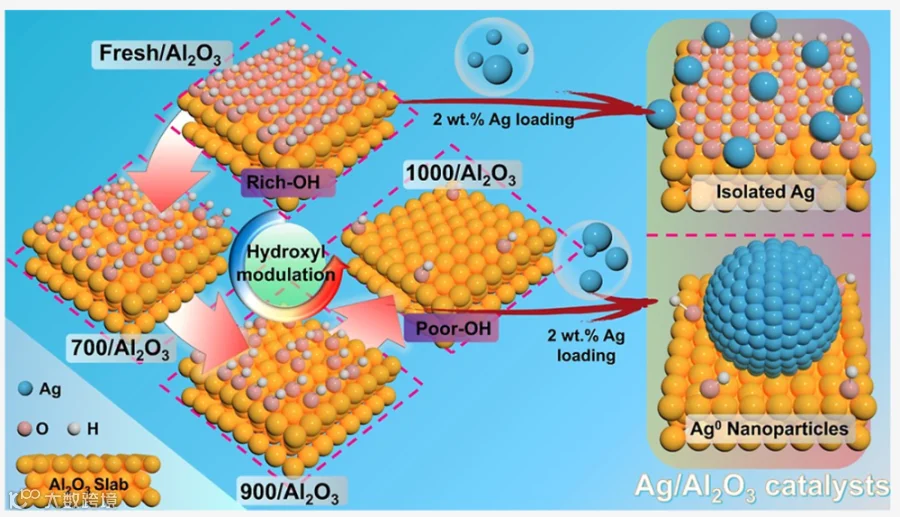
调控催化剂中活性中心的尺寸对其活性和选择性具有深远影响。本文展示了一种在Al2O3载体上精确控制银物种尺寸的策略,使2%Ag/Al2O3催化剂能够针对不同催化反应进行定制化设计。基于羟基作为银锚定位点的机理,我们合成了富含羟基的Al2O3载体,随后通过焙烧调控羟基含量。该方法使银物种能够以孤立原子、团簇或纳米颗粒等形式存在于Al2O3表面。随着银尺寸从单原子逐渐增大至团簇和纳米颗粒,催化剂的氧气活化能力逐步增强,从而导致涉氧反应性能的显著变化。值得关注的是,这种富羟基载体的合成方法简便且具有良好重现性。我们的研究结果为合理设计具有可控活性中心尺寸的负载型银催化剂提供了重要见解,适用于多种催化应用场景。
氧化铝负载银(Ag/Al2O₃)催化剂在工业和环境催化领域的重要性日益凸显,这源于其在基础研究和实际应用中的巨大潜力。这类催化剂尤为显著的特征是其明显的尺寸依赖性效应——银物种的尺寸对催化性能具有决定性影响。例如Lei等研究表明,在直接丙烯环氧化反应中,配位数较低的亚纳米银簇(约0.4 nm)比传统较大尺寸的纳米银颗粒(>3 nm)表现出显著优越的催化活性,这种活性增强主要归因于亚纳米簇中d带中心的上移。相反,对于CO氧化等反应,较大的银纳米颗粒(AgNPs)(约10 nm)作为主要活性位点,其优越的氧活化能力促进了表面活性原子氧的生成,显著提升了催化活性。此外,单原子催化剂因具有最大化的原子利用效率近年来受到广泛关注。这些发现突显了金属物种尺寸对催化剂电子结构、表面形貌及金属-载体相互作用的深远影响,进而影响催化活性和选择性。
2、单原子到团簇乃至纳米颗粒的金属尺寸如何精确控制?
鉴于这种尺寸依赖性行为,实现从单原子到团簇乃至纳米颗粒的金属尺寸精确控制,对于优化催化剂性能和阐明反应机制至关重要。当前负载型催化剂的尺寸控制主要通过原子层沉积(ALD)和金属负载量调控等方法实现。其中ALD凭借其三维共形包覆和原子级精度优势,在多相催化领域展现出巨大应用前景。但该方法所需的高精度设备和昂贵前驱体严重制约了其大规模应用的扩展性。通过金属负载量调控实现尺寸控制的方法更为简便易行,因而成为负载型催化剂的常用策略。然而这种方法通常会导致金属利用率降低,特别是当团簇和颗粒作为活性位点的贵金属催化剂时,会大幅增加催化剂成本。因此亟需开发简单、精准且经济高效的调控银活性位点尺寸以满足实际应用需求。
前期研究表明,Al₂O₃表面的末端羟基可作为银原子的锚定位点。羟基的丰度决定了银的分散状态:高密度羟基促进原子级分散的银,而低密度则导致银聚集成团簇或纳米颗粒。因此,通过调控羟基数量可实现银锚定状态与尺寸的精确控制。本研究采用化学沉淀法合成富羟基Al₂O₃,并通过改变焙烧温度调节表面羟基含量,从而实现了银从单原子到团簇乃至纳米颗粒的尺寸调控,进而通过不同探针反应研究了结构-性能关系。本研究以银在Al₂O₃表面的锚定机制为理论框架,为理性设计高性能Ag/Al₂O₃催化剂开辟了新途径。
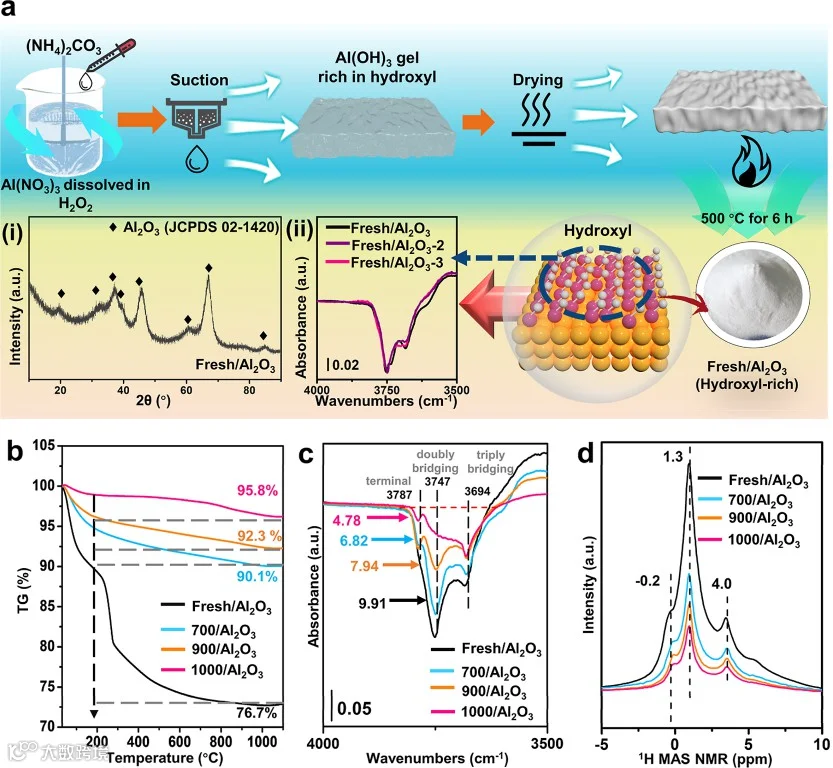
采用化学沉淀法制备了Al2O3载体。将硝酸铝溶于50 vol.% H2O2溶液中持续搅拌至溶液澄清,在搅拌状态下滴加浓度为1 mol/L的(NH4)2CO3水溶液,将溶液pH值调节至8-9之间。使悬浮液在50℃下经真空旋转蒸发仪处理30分钟,随后于105℃干燥24小时并在500℃下煅烧6小时。最后将所得样品加入去离子水中超声清洗15分钟后过滤,重复三次该流程后,样品于105°C干燥12小时,最终获得富含羟基的Al2O3载体。通过不同焙烧温度来调控载体的羟基浓度。
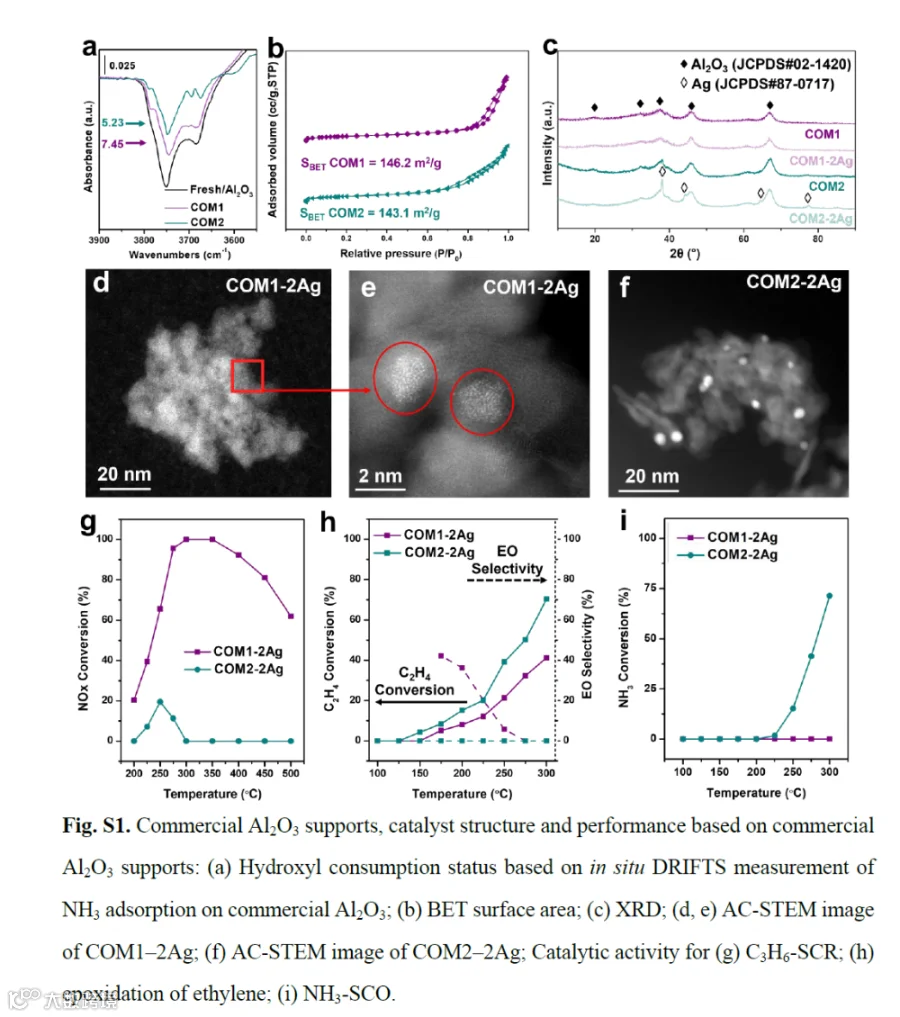
采用湿式浸渍法合成催化剂。将Ag负载于预先合成的Al2O3载体上:首先将适量Fresh/Al2O3、700/Al2O3、900/Al2O3或1000/Al2O3在搅拌条件下加入去离子水中形成悬浮液,随后加入AgNO3水溶液。浸渍3小时后,通过真空旋转蒸发仪对悬浮液进行干燥处理。样品于105°C下干燥12小时,随后在500°C空气中煅烧3小时,并通过40-60目筛网进行筛分。Al2O3上的Ag负载量控制在2 wt.%。本研究用于对比实验的商业γ-Al2O3样品(Sigma-Aldrich,平均粒径=10 nm)命名为COM1和COM2,以其为载体负载2 wt.% Ag合成的催化剂分别命名为COM1−2Ag和COM2−2Ag。需要说明的是,采购的两批商业γ-Al2O3具有相同的规格参数和晶相特征。
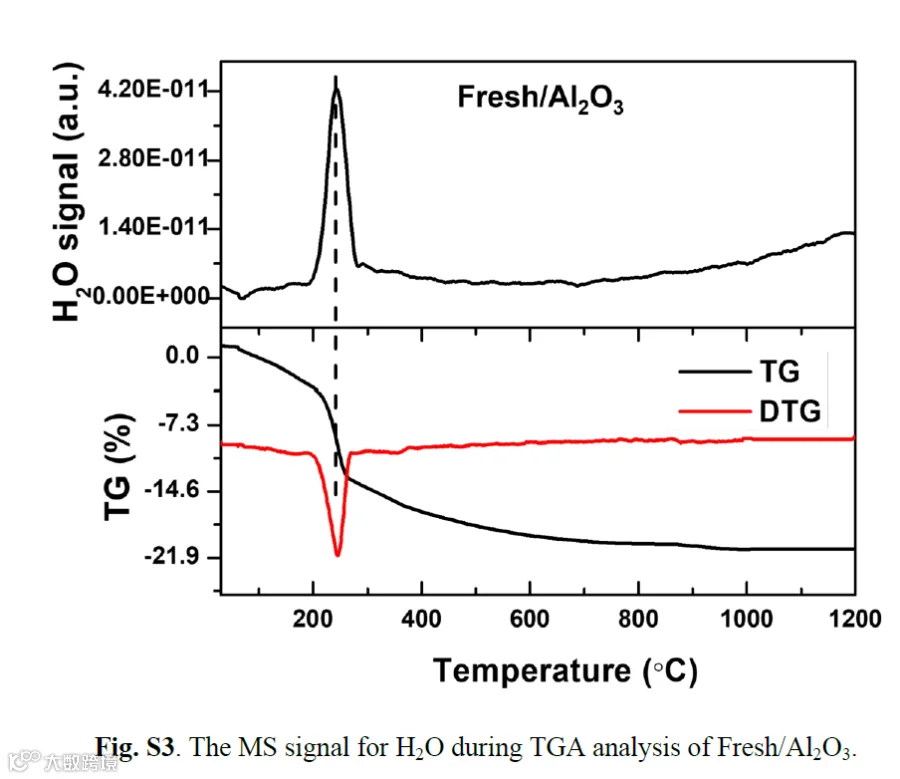
已有充分研究证实,催化剂表面的羟基在高温下会以水分子形式脱除。热重分析(TGA)为探究催化剂及其载体表面羟基提供了重要依据。如图S3所示,当温度超过200°C时,Fresh/Al2O3表面吸附水和羟基被同步脱除,导致其出现显著质量损失。在1000°C时,Fresh/Al2O3、700/Al2O3、900/Al2O3和1000/Al2O3的质量分别降至初始值的76.7%、90.1%、92.3%和95.8%(图1b)。NH3吸附原位漫反射傅里叶变换红外光谱(DRIFTS)进一步为样品表面羟基含量提供了间接测定(图1c)。在3787、3747和3694 cm−1处观察到的Al2O3样品负峰对应于分别为末端羟基、双桥羟基和三桥羟基。
Figure 1. Schematic diagram of the preparation of hydroxyl-rich Al2O3 and determination of hydroxyl content. (a) Hydroxyl-rich Al2O3 preparation procedure , XRD, and comparison of hydroxyl content of Fresh/Al2O3 synthesized in three repeated processes; (b) thermogravimetric analysis (TGA); (c) OH consumption status based on in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurement of NH3 adsorption on Al2O3; (d) normalized proton magic-angle spinning nuclear magnetic resonance (1H MAS NMR) of the Al2O3 samples.
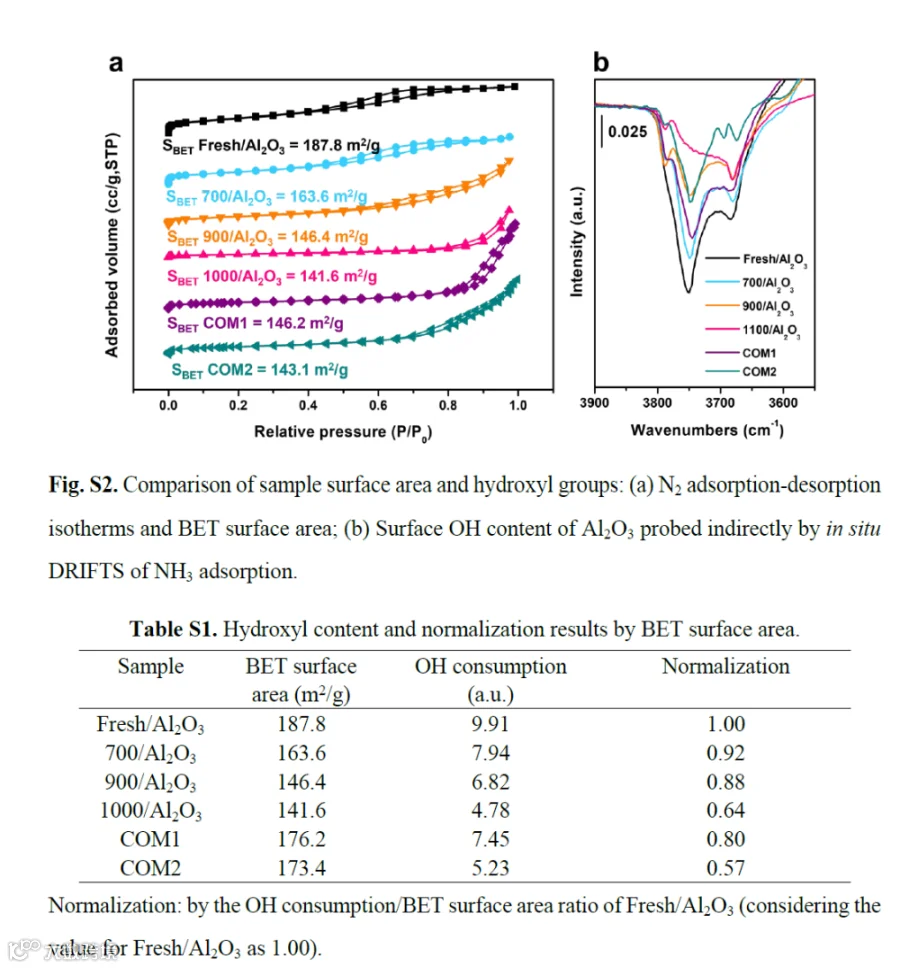
随着Fresh/Al₂O₃样品煅烧温度的逐步升高,羟基含量呈现递减趋势。Fresh/Al₂O₃、700/Al₂O₃、900/Al₂O₃及1000/Al₂O₃的羟基峰面积依次为9.91、7.94、6.82和4.79 au·cm⁻¹,这表明二次煅烧能有效且定量地去除Fresh/Al₂O₃表面的羟基基团。通过¹H MAS NMR进一步验证了羟基含量与煅烧温度的关系(图1d)。¹H MAS NMR谱图显示出三个主要特征峰:-0.2 ppm处的峰归属于四配位铝结合的末端羟基(HO-μ₁-Alᴵⱽ),1.3 ppm处的峰对应于五配位铝及n配位铝结合的双桥羟基(HO-μ₂-Alⱽ, HO-μ₂-Alⁿ),而4.0 ppm处的宽谱带则源于n配位铝结合的三桥羟基(HO-μ₃-Alⁿ)及残留水分。结果表明随着煅烧温度升高,所有类型的表面羟基均出现减少,其中1.3 ppm处的双桥羟基减少最为显著。这些发现与热重分析(TGA)和原位漫反射傅里叶变换红外光谱(DRIFTS)实验结果一致。过高的煅烧温度可能导致Al₂O₃结构坍塌,使其比表面积减小并降低羟基含量(图S2),这可能会影响活性位点的分散度。因此,本研究将合成Al₂O₃与商业Al₂O₃进行对比,并根据比表面积对羟基含量(源自原位DRIFTS结果)进行标准化处理。标准化结果如表S1所示,表明单位面积表面羟基含量遵循Fresh/Al₂O₃ (1.00) > 700/Al₂O₃ (0.92) > 900/Al₂O₃ (0.88) > 1000/Al₂O₃ (0.64)的规律。由此可见,羟基含量并非仅由比表面积决定。合成的Fresh/Al₂O₃展现出较高的单位面积羟基含量,证明了进一步煅烧调控羟基含量的有效性。
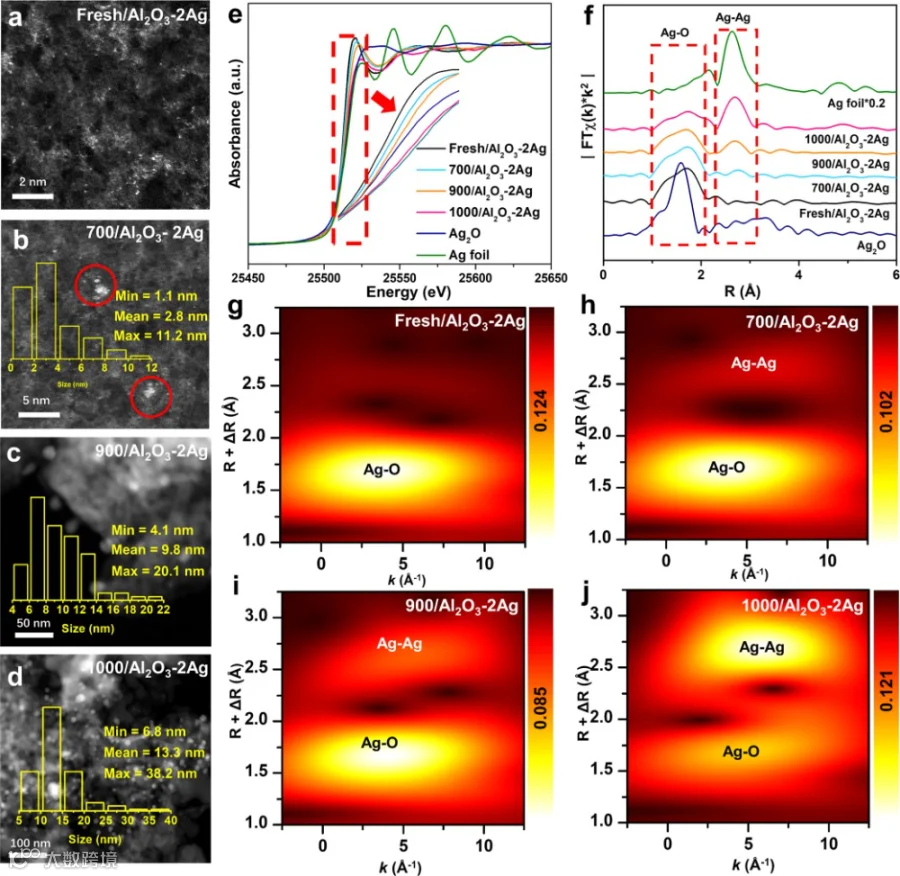
与单一Al₂O₃载体表面的羟基含量相比,当负载2 wt.%银后,末端和桥接羟基被部分消耗,表明银原子锚定在Al₂O₃表面的羟基上(图S4和S5)。此外,随着羟基含量降低,可用于锚定位点的羟基数量减少可能导致银团聚形成银纳米颗粒(AgNPs)。X射线光电子能谱(XPS)结果进一步证明了银物种对羟基占据的有效性(图S6和表S2)。为验证2 wt.%银负载后羟基峰强度降低是由于银消耗羟基所致,新一组设计了相关实验。将Fresh/Al2O3分散于去离子水中形成悬浮液,搅拌2小时后进行真空旋转蒸发,105°C下干燥,最后在500°C下煅烧。结果表明这些处理对Al2O3载体的羟基浓度影响甚微(图S7),说明银负载后观测到的羟基峰面积减小确实是由银与表面羟基的锚定作用所致。高角度环形暗场扫描透射电子显微镜(HAADF-STEM)清晰展现了银在不同羟基含量Al2O3表面的分散状态(图2a-d)。Fresh/Al2O3−2Ag呈现出强锚定的孤立银单原子,而700/Al2O3−2Ag则存在银团簇。在900/Al2O3−2Ag和1000/Al2O3−2Ag表面分别出现了平均粒径为9.8 nm和13.3 nm的大尺寸银纳米颗粒。这表明通过调控Al2O3表面羟基可有效控制活性银金属的分散状态。如图2e所示归一化银K边XANES谱表明,700/Al2O3−2Ag、900/Al2O3−2Ag和1000/Al2O3−2Ag催化剂中的银物种以Ag0和Ag+混合态存在;而Fresh/Al2O3−2Ag中的银物种价态主要以Ag+为主。傅里叶变换银K边EXAFS谱(图2f)进一步证实该结论:银箔及所有催化剂在2.67 Å处(未相位校正)均出现对应Ag-Ag金属键的特征峰,且Ag-Ag配位壳层强度随Al2O3载体羟基含量降低而增强。EXAFS曲线拟合提供了不同催化剂中银的定量结构参数(图S8)。
Figure 2. State of the catalyst active center. (a−d) HAADF-STEM images and Ag size distributions; (e) Ag−K edge X-ray absorption near edge structure (XANES); (f) Ag−K edge extended X-ray absorption fine structure spectroscopy (EXAFS); (g−j) Wavelet transform (WT) of Ag−K edge for different catalysts.
经同步辐射吸收精细谱XAS测试Fresh/Al2O3−2Ag、700/Al2O3−2Ag、900/Al2O3−2Ag和1000/Al2O3−2Ag的Ag-Ag键配位数分别约为0.1、0.4、1.0和2.4(表S3)。为更精确区分银原子分散状态,对k2加权EXAFS信号进行小波变换(WT)分析。二维WT图谱如图2g-j所示:Fresh/Al2O3−2Ag的WT强度最大值出现在1.5-2.0 Å处(Ag-O配位壳层),而在2.2-3.0 Å处的Ag-Ag配位壳层几乎不可见(图2g);700/Al2O3−2Ag样品呈现较弱的Ag-Ag配位和较低的Ag-O强度,表明存在银团簇或亚纳米颗粒;1000/Al2O3−2Ag样品则显示出最强的Ag-Ag配位强度和最弱的Ag-O配位。这些结果与X射线衍射(XRD)、紫外-可见漫反射光谱(UV-vis DRS)及X射线光电子能谱(XPS)分析结果一致(图S9)。
Al³⁺五配位结构(Al3+penta)一直被视为图4表面活性金属原子的锚定位点之一。根据Al魔角旋转核磁共振谱(MAS NMR)分析结果,载体经二次煅烧后其Al³⁺五配位结构未发生显著变化(图S10)。这表明Al³⁺五配位结构并非本研究影响银分散度的关键因素。因此,通过调控Al₂O₃载体表面羟基基团,可显著影响负载银物种的粒径尺寸与价态分布,从而可能从根本上改变催化剂性能表现。
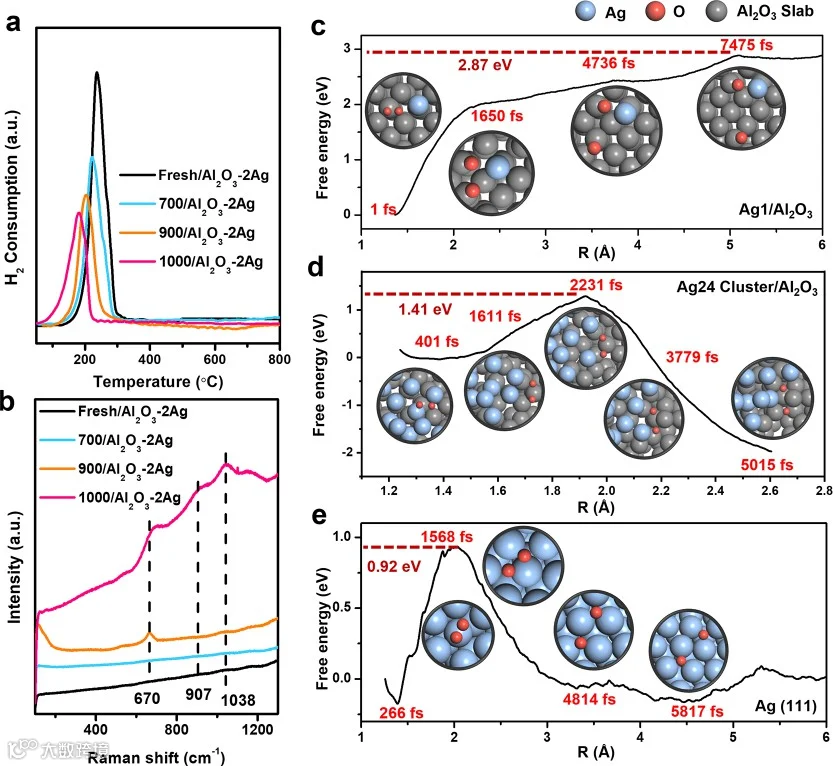
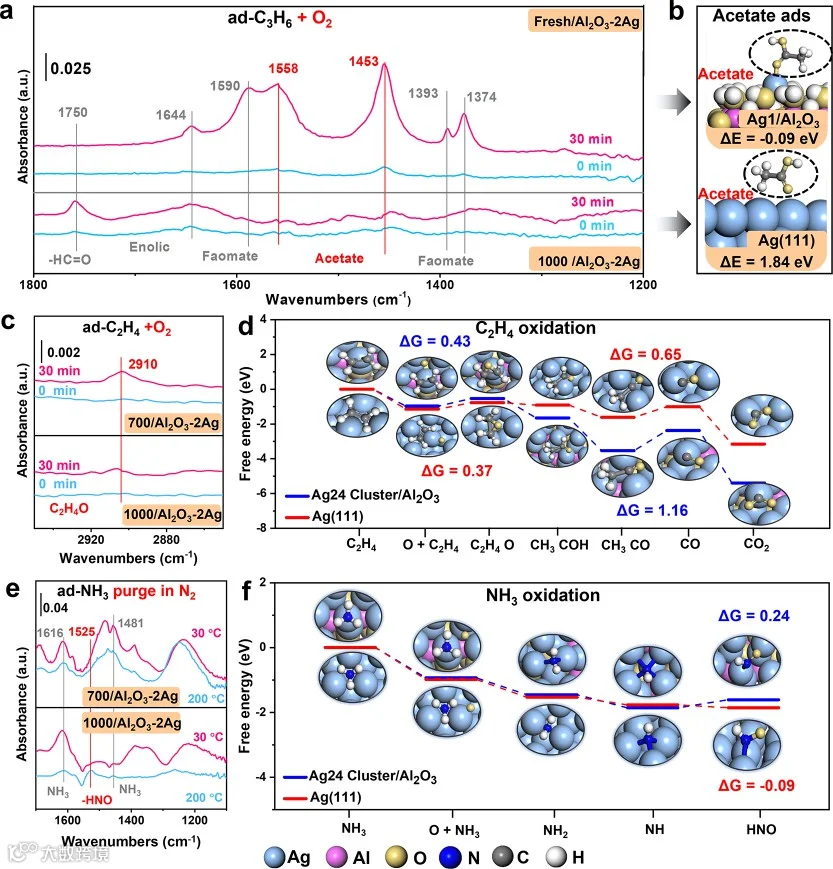
采用氢气程序升温还原(H2-TPR)技术测定催化剂的氧化还原性能。如图3a所示,Fresh/Al2O3−2Ag、700/Al2O3−2Ag、900/Al2O3−2Ag和1000/Al2O3−2Ag样品分别在237、225、199和175°C处出现明显的H2消耗峰,表明随着银颗粒尺寸增大,其氧化还原性能逐步增强。通过原位拉曼光谱观测了这些样品表面氧物种的形态(图3b):670 cm−1处的特征峰归属于原子态氧(O*),907和1038 cm−1处的特征峰则归属于分子态氧(O2*)(特征峰归属分析见图S11和S12)。Fresh/Al2O3−2Ag和700/Al2O3−2Ag未检测到氧物种相关特征峰,表明这两种样品对O2的吸附、活化及解离能力较弱。900/Al2O3−2Ag在670 cm−1处出现原子态氧(O*)特征峰,而907和1038 cm−1处的分子态氧(O2*)信号显著减弱,说明该催化剂表面对O2的吸附能力有限。1000/Al2O3−2Ag表现出更优的氧活化能力,其O2*特征峰强度最高。较大尺寸的银纳米颗粒(Ag NPs)在低温下始终展现出更强的O2分子吸附与活化能力。此外,这些催化剂在高温条件下对O2分子的分解和活化能力也显著增强(图S13)。通过监测O2分子中两个氧原子间距离的变化,获得了Ag1单原子、Ag24团簇和Ag(111)模型表面在长期模拟过程中的能垒(图3c−e)。所有经退火处理和NVT-1 fs结构优化的模型见图S14。从头算分子动力学(AIMD)模拟揭示了O2在Ag1/Al2O3表面从1至7475 fs的解离路径:O-O键从吸附态初始断裂及两个氧原子迁移5.2 Å的过程均为吸热反应,需消耗2.87 eV能量。对于Ag24 Cluster/Al2O3,O-O键在2231 fs处断裂是整个O2分解过程中能量需求最高的步骤,需1.41 eV能量;随后氧原子的迁移为自发过程。而Ag(111)表面的O2分解仅在1568 fs处需0.92 eV能量。
Figure 3. O2 activation capacity of catalysts. (a) H2-TPR; (b) in situ Raman spectra at 275 °C (test conditions: 10 vol.% O2/N2); (c−e) snapshots ofthetrajectoryO2activationonAg1/Al2O3,Ag24Cluster/Al2O3andAg(111)at275°C(thesimulationresultscorrespondtothemodelsshownin the insets, with the horizontal coordinate (Å) representing the distance between two O atoms)
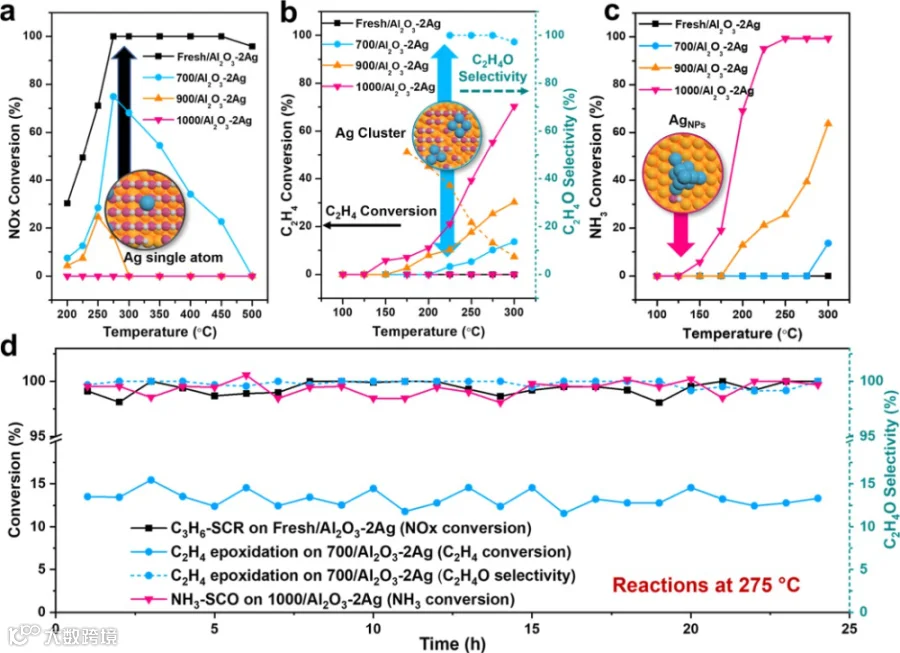
银单原子、团簇和纳米颗粒催化剂在不同反应中表现出独特的活性和选择性(图4)。在C3H6−SCR反应中,Fresh/Al2O3−2Ag是最有效的催化剂,在275至450°C温度区间内实现氮氧化物完全转化(图4a)。随着银活性中心尺寸增大,NOx转化率(XNOx)逐渐降低。700/Al2O3−2Ag和900/Al2O3−2Ag催化剂呈现火山型曲线,其最高XNOx分别出现在275和250°C。1000/Al2O3−2Ag样品无催化活性,可能因其过强的氧化能力导致C3H6被完全氧化。在乙烯环氧化反应中,银团簇催化剂(700/Al2O3−2Ag)相较于单原子和纳米颗粒催化剂展现出显著优势,实现了接近100%的环氧乙烷(C2H4O)选择性(图4b)。较大银颗粒更强的氧化能力导致乙烯被完全氧化为CO2。与上述反应不同,银纳米颗粒催化剂在NH3−SCO反应中表现最优。负载于Al2O3上的银纳米颗粒分解O2为氧原子的低能垒是促进NH3氧化的关键因素,而原子态氧与NH3的反应也被认为是NH3氧化的关键步骤。通过275°C下24小时连续反应进一步验证了样品在各自特征反应中的稳定性(图4d)。所有催化剂的活性和选择性在24小时内均未出现显著下降。
Figure 4. Catalyst activity test results. (a) C3H6−SCR (800 ppm of NO, 1600 ppm of C3H6, 10 vol.% O2, 1 vol.% H2, gas hourly space velocity (GHSV) = 120,000 h−1); (b) C2H4 epoxidation (5 vol.% C2H4, 5 vol.% O2, GHSV = 8000 h−1); (c) NH3−SCO (500 ppm of NH3, 10 vol.% O2, GHSV = 120,000 h−1); (d) stability of Fresh/Al2O3−2Ag, 700/Al2O3−2Ag, and 1000/Al2O3−2Ag catalysts in characteristic reactions at 275 °C.
值得一提的是,在众多催化剂中,唯有Fresh/Al₂O₃−2Ag催化剂中的银物种呈现出单一化学态价态。虽然银价态存在一定影响,但决定催化性能的主要因素仍是银物种的粒径尺寸。此外,Fresh/Al₂O₃−2Ag、700/Al₂O₃−2Ag和1000/Al₂O₃−2Ag分别在C₃H₆−SCR、C₂H₄环氧化和NH₃−SCO反应中循环使用三次后,活性未出现显著下降(图S23)。这表明通过锚定位点调控合成的单原子、团簇和纳米粒子催化剂具有优异的催化稳定性。进一步分析证实,在275°C反应24小时后,Fresh/Al₂O₃−2Ag、700/Al₂O₃−2Ag和1000/Al₂O₃−2Ag中的银物种分别保持单原子态、团簇态及约16纳米的银纳米粒子态,且氧化态未发生显著变化(图S24-S26)。这些结果进一步证明了通过简单调控Al₂O₃载体表面羟基含量所制备的不同尺寸银活性中心具有卓越的稳定性。
5、调控Al₂O₃载体表面羟基浓度来实现粒径控制普适性
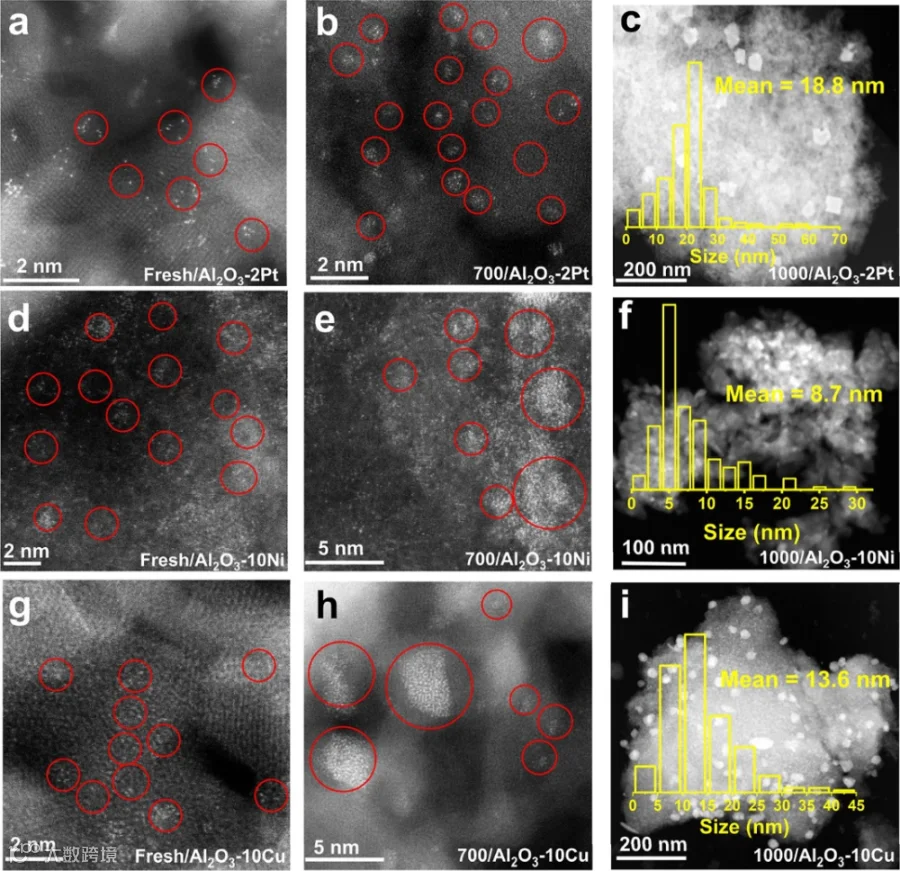
Pt、Ni、Cu等金属原子同样以末端羟基和双配位桥接羟基作为锚定位点。为验证该调控方法的普适性,我们将多种金属原子沉积至Fresh/Al₂O₃、700/Al₂O₃及1000/Al₂O₃载体上,并系统分析了这些样品中金属物种的分散状态。X射线衍射(XRD)结果表明:在Fresh/Al₂O₃−2Pt、700/Al₂O₃−2Pt、Fresh/Al₂O₃−10Ni、700/Al₂O₃−10Ni、Fresh/Al₂O₃−10Cu及700/Al₂O₃−10Cu样品中,均未观察到与Pt、Ni或Cu物种相关的衍射峰(图S37),说明这些样品中的金属原子呈高度分散状态。HAADF-STEM分析进一步佐证了XRD的发现。在Fresh/Al₂O₃−2Pt样品中,铂原子主要以单原子形式分散在Al₂O₃表面。700/Al₂O₃−2Pt样品中的铂物种则主要形成5-20个原子的团簇,仅存少量单原子态铂。而在1000/Al₂O₃−2Pt样品中观察到平均粒径为18.8纳米的铂颗粒(图6a−c)。Fresh/Al₂O₃−10Ni中的镍原子主要呈单原子态分布;700/Al₂O₃−10Ni中的镍原子虽未形成规整的NiO纳米颗粒,但已在Al₂O₃表面发生团聚形成团簇;1000/Al₂O₃−10Ni中则可见平均粒径8.7纳米的NiO纳米颗粒(图6d−f)。Fresh/Al₂O₃−10Cu中的铜主要以单原子形式分散,伴有少量团簇;700/Al₂O₃−10Cu中的铜原子团聚程度加剧,形成不规则CuO团簇;1000/Al₂O₃−10Cu中可见平均粒径约13.6纳米的CuO物种均匀分布于Al₂O₃表面(图6g−i)。这些结果证实,通过调控Al₂O₃载体表面羟基含量来设计金属活性中心尺寸的方法具有普适性。
Figure 5. Key in situ DRIFTS and DFT calculation results for various reactions on Ag single atom, cluster, and nanoparticle catalysts. (a,b) In situ
DRIFTS of preadsorbed C3H6 oxidation at 275 °C on Fresh/Al2O3−2Ag and 1000/Al2O3−2Ag surfaces, and the energy of acetate adsorption on
Ag1/Al2O3 and Ag(111) surfaces; (c,d) in situ DRIFTS of preadsorbed C2H4 oxidation at 275 °C on 700/Al2O3−2Ag and 1000/Al2O3−2Ag
surfaces, and energy barrier of C2H4 oxidation on Ag24 Cluster/Al2O3 and Ag(111); (e,f) in situ DRIFTS of adsorbed NH3 at 30 °C, followed by
desorption at 200 °C on 700/Al2O3−2Ag and 1000/Al2O3−2Ag surfaces, and energy barrier of NH3 oxidation on Ag24 Cluster/Al2O3 and
简言之,随着银活性中心尺寸从单原子增大至团簇和纳米颗粒,其对氧气的活化能力相应增强。这一现象显著影响了2%Ag/Al₂O₃催化剂在多种反应中的性能表现。在C₃H₆选择性催化还原(SCR)和C₂H₄环氧化反应中,过大的银纳米颗粒(AgNPs)因其过强的氧气活化能力,易将中间产物(即乙酸酸盐物种和C₂H₄O)过度氧化为CO₂,从而导致氮氧化物转化率(XNOx)和环氧乙烷产率(C₂H₄O)下降。相反,NH₃选择性催化氧化(SCO)反应需要高活性氧原子与NH₃反应,表明较大尺寸的AgNPs更有利于提升氨转化率(XNH₃)。需特别指出的是,银物种在Al₂O₃上的分散度受羟基丰度影响——末端羟基与双配位桥接羟基作为银原子的锚定位点。这表明其他同样以羟基为锚定位点的金属,亦可通过调控Al₂O₃载体表面羟基浓度来实现粒径控制。
Figure 6. HAADF-STEM images of catalysts. (a−c) Fresh/Al2O3−2Pt, 700/Al2O3−2Pt and 1000/Al2O3−2Pt; (d−f) Fresh/Al2O3−10Ni, 700/ Al2O3−10Ni and 1000/Al2O3−10Ni; (g−i) Fresh/Al2O3−10Cu, 700/Al2O3−10Cu and 1000/Al2O3−10Cu.
成功合成了富含羟基且能够锚定银原子的氧化铝载体。通过煅烧调节羟基含量,在不改变银负载量的前提下,设计出从单原子到团簇再到纳米颗粒的不同尺寸银活性中心。随着银活性中心尺寸从单原子逐步增大至团簇和纳米颗粒,其氧气活化能力逐渐增强。这导致银单原子在C3H6选择性催化还原反应中表现出优异活性,银团簇在乙烯环氧化反应中呈现卓越的环氧乙烷选择性,而银纳米颗粒更倾向于氨选择性催化氧化反应。通过这种羟基调控策略,研究团队还成功制备出铂、镍和铜的单原子、团簇及纳米颗粒催化剂。预计通过控制氧化物载体表面羟基含量,可为开发成本效益高且性能优异的催化剂提供新策略。此外,这种催化剂锚定位点设计的新方法为实用型催化剂的研发提供了更具创新性的视角。
免责声明:所载内容、部分引用图片、表格来源于互联网,微信公众号等公开渠道,我们对文中观点持中立态度,本文仅供参考、交流。转载的稿件及图片、字体等版权归原作者和机构所有,如有侵权,请联系我们删除。