


高效液相色谱法系采用高压输液泵将规定的流动相泵入装有填充剂的色谱柱,对供试品进行分离测定的色谱方法。注入的供试品,由流动相带入色谱柱内,各组分在柱内被分离,并进入检测器检测,由积分仪或数据处理系统记录和处理色谱信号。高效液相色谱法收载于《中国药典》2020年版四部通则,在药物含量、有关物质及组分等的检测中广泛应用。本次讨论为您解锁液相色谱分析方法开发的奥秘。
液相色谱分析方法的选择至关重要,影响药物质量分析时结论的可靠性。在进行液相色谱分析方法开发时,需调整方法中的各个参数,以保证分析方法的专属性、准确度、精密度等。为优化分析方法,通常调整的参数有:色谱柱、流动相、流速、溶剂、进样体积、柱温等。
色谱柱是色谱系统的心脏,担任分离物质的重要作用,是否选择合适的色谱柱对物质的分离起到决定性作用。进行方法开发时,需根据待分离物质的结构上的差异,选择正确的色谱柱。如待分离物质为疏水性物质,且疏水性存在差异通常选择C18、C8柱;待分离物质极性较大,在C18、C8柱上未保留,可选择HILIC柱;待分离物质为手性异构体,根据其化合物结构,可选择相应手性分离色谱柱,如Chiral-V、Chiral-T等。
选择色谱柱时,还需要根据所用流动相的pH,选择满足相应pH范围的色谱柱,如EC-C18的pH范围为2.0-8.0,SB-C18的pH范围为1.0-8.0,HPH-C18的pH范围为3.0-11.0。同时,应该关注是否封端,若未封端,则在流动相中应考虑加三氟乙酸、三乙胺等进行扫尾,或者使用甲醇作为有机相等。
表1 不同填料色谱柱信息
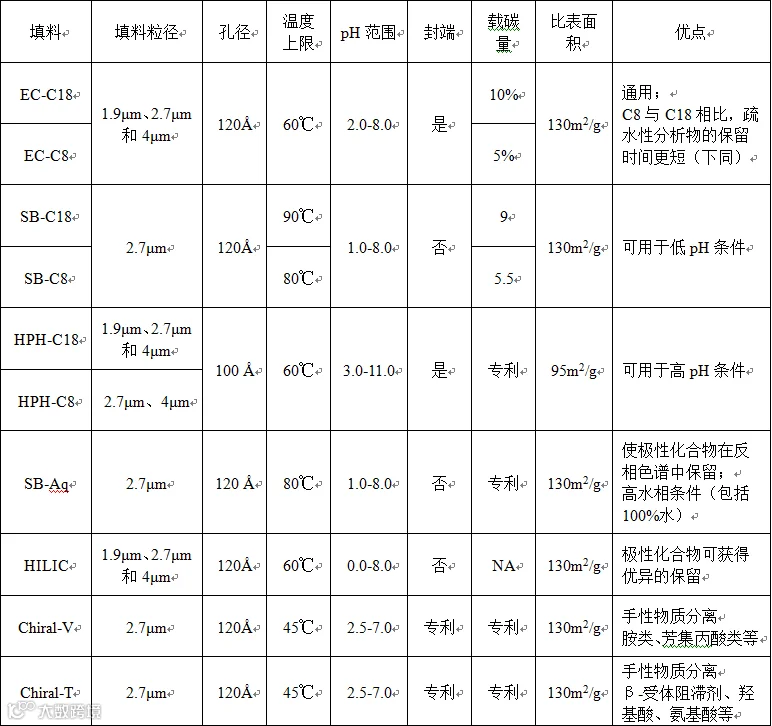
不同厂家色谱柱工艺不同,导致其质量存在一定差异,使用某一厂家的色谱柱开发出方法后,需对比不同厂家色谱柱检测结果的差异,若未对比或者对比后存在差异,则方法中需明确规定所用色谱柱的厂家。表1中所列色谱柱信息由安捷伦提供,上述举例的色谱柱数据均为安捷伦色谱柱的数据。
➢流动相比例
液相色谱分为正相色谱及反相色谱,正相色谱中固定相极性大于流动相极性,用于分离极性物质,极性小的先流出;反相色谱中流动相极性大于固定相,用于分析非极性及弱极性的物质,极性大的先流出。
正相色谱系统的流动相常用两种或两种以上的有机溶剂,如二氯甲烷和正己烷等,反相色谱系统的流动相常用甲醇-水系统或乙腈-水系统。在选择流动相比例时,可以采用将高洗脱能力的溶剂比例由低逐渐升高的方式,选择合适的流动相比例,如反相色谱中的甲醇-水系统,甲醇的极性比水小,故甲醇洗脱能力比水强,则将甲醇比低比例(如5%,若使用耐水柱如SB-Aq,则可从0%开始)慢慢升至100%,相应水相的比例由高比例慢慢降低。根据待分析物质出峰时的流动相比例,确定方法中的流动相比例。
➢洗脱程序
洗脱方式分为等度洗脱和梯度洗脱。等度洗脱是指在同一个分析周期中,流动相的浓度配比不发生变化;梯度洗脱是指在同一个分析周期中,按一定程度不断改变流动相的浓度配比。
等度洗脱相较于梯度洗脱对仪器的要求更低,基线更稳定,但在分离杂质和主成分或杂质和杂质时,若两者极性相差较大,会导致极性小的物质保留时间太长,出峰晚,使其色谱峰太宽且分析时间太长。这种情况采用梯度洗脱就很有必要。分别对不同物质选择合适的流动相比例,设定洗脱程序,使其在不同比例处进行分离。
设定梯度时,宜选择洗脱能力弱的比例在前,洗脱能力强的比例在后,否则待分离的物质中易洗脱的物质可能不保留。如在反相色谱中,使用乙腈-水系统作为流动相,乙腈的极性比水小,即洗脱能力比水强。待分离物质A、B均为弱极性物质,其中A的极性比B小,A在乙腈:水(50:50)时开始被洗脱,B在乙腈:水(10:90)开始被洗脱,若洗脱程序开始的比例为乙腈:水(50:50),而B比A更易被洗脱,则在乙腈:水(50:50)下几乎不保留。
➢有机相类型
不同溶剂的极性及紫外截止吸收波长存在一定差异,正相色谱系统的流动相为极性小的溶剂,如二氯甲烷和正己烷;反相色谱系统的流动相为极性大的溶剂,通常为有机相及水相的混合系统,其中有机相常用的溶剂为甲醇或乙腈。当选择紫外末端波长检测时,由于甲醇存在末端吸收,优先选择乙腈体系;在使用未封端的色谱柱且流动相的pH为酸性(如pH为3.5或更低时)的情况下,对碱性物质进行分离时,若使用乙腈出现拖尾的情况,则可改用甲醇,使其与色谱柱上的硅醇基结合,避免拖尾;当使用的方法系统压力太大时,可以尝试用乙腈替代甲醇,由于乙腈粘度比甲醇小,产生的系统压力更小。
表2 不同溶剂信息表
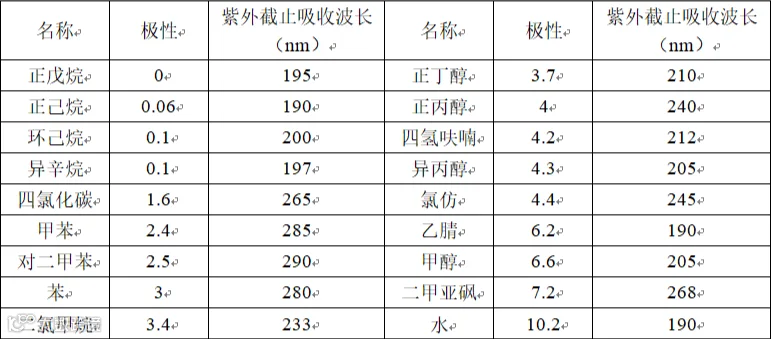
若所选定的流动相pH接近物质的pKa值,则离子态与分子态共存,而离子态与分子态亲水性存在差异,会导致其保留行为出现差异,使其出现拖尾、前沿、叉峰、双峰等情况。通常认为两种状态的浓度之比大于100时,该溶液中只以一种状态存在,另一种状态可忽略不计,即当|pH-pKa|大于2时,化合物只以离子态或分子态存在。故流动相pH值不应选择pKa-2~pKa+2范围内,而pH>pKa+2时,酸性物质只以离子态存在,碱性物质只以分子态存在,pH<pKa-2时,相反。
➢缓冲盐类型及浓度
由于待测的供试品溶液存在一定的酸碱性,进样后易导致局部流动相的pH值发生变化,影响其分离,故通常在流动相中使用缓冲盐。选择缓冲盐时,需根据所需流动相pH值选择合适的缓冲体系。缓冲溶液有缓冲作用的pH范围约为pKa±1范围内,其中pH=pKa时,缓冲能力最强,不同缓冲盐pKa及缓冲范围见表3。使用缓冲溶液时,应尽可能使用低浓度缓冲盐。但缓冲盐浓度太低,缓冲能力不够,而浓度太高,在与有机相混合后容易在系统中析出,导致仪器或色谱柱堵塞,所以浓度的选择也十分重要。
表3 常用的缓冲溶液
名称 |
pKa |
缓冲范围 |
乙酸盐(铵盐) |
4.76 |
3.76~5.76 |
甲酸盐(铵盐) |
3.75 |
2.75~4.75 |
磷酸盐 |
2.15(pKa1) |
1.15~3.15 |
7.2(pKa2) |
6.2~8.2 |
|
12.3(pKa3) |
11.3~13.3 |
|
乙酸铵 |
9.2 |
8.2~10.2 |
➢离子对试剂
当待分析物质在流动相中为离子态时,其在反相色谱柱上的保留时间很短或者根本不保留,这时要加入相应的离子对试剂,将待分析物质上的离子进行结合,形成在柱子上有保留的分子。用于酸性化合物的离子对试剂四丁基氢氧化铵(10%水溶液)、四丁基溴化铵等;用于碱性化合物的离子对试剂有庚烷磺酸钠、辛烷磺酸钠等。
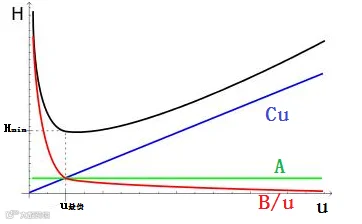
根据Van Deemter方程H=A+B/u+Cu,塔板高度与流动相的线速度有关,而塔板数N=L/H, 色谱柱长度L确定后,塔板高度越小,塔板数越大,柱效越好。不同填料、粒径、不同孔径,最佳流速均不一致,需分别进行确定。其中同一填料、不同色谱柱间,最佳流速的转换公式为F2=F1×[(dc22×dp1)/(dc12×dp2)](其中dc为色谱柱内径,dp为色谱柱粒径)。
用于溶解或稀释样品的溶剂,通常选择与初始流动相相同的溶液,避免出现溶解度不同导致样品析出或pH、极性差异太大导致出现溶剂效应使峰形较差等。但如果以流动相作为溶剂出现溶液稳定性差、无法溶解等情况,也可以另外选择合适的溶液作为溶剂,但需确保不会出现上述问题。
样品的进样体积首先需保证在仪器进样器的准确抽液范围内,在样品浓度确定的情况下,需保证样品量不得过大使其超载。若所用溶剂与流动相差异较大,为避免溶剂效应,通常不使用较大的进样体积。
在改变流动相无法分离两个组分时,通过调整柱温可能会有意想不到的效果。通常,升高温度会减小化合物的保留,但升高或降低温度对不同物质的保留因子影响情况不同,可能导致两个物质间的选择度增大。如杂质1和杂质2,其原保留因子分别为k1、k2(k2>k1,即杂质1先出峰),两个杂质的选择度α= k2/ k1,当减低温度时,两个杂质保留因子均增大,其中对杂质2影响更大,杂质2保留因子由k2增大到1.2 k2,而杂质1增大到1.1k1,则降温后的α´大于α,两个杂质选择度更高,分离度更好。
不同物质根据其不同性质选择不同检测器,其中,有紫外或可见光吸收的物质,可使用紫外-可见分光检测器。在选择检测波长时,宜选择有最大吸收的波长作为检测波长,主要是吸光度大,其检测灵敏度更高。
但检测有关物质时,由于待检测的物质较多,不同物质其最大吸收波长均有所差异,则需进行综合考虑。若无合适的波长,则需考虑设定不同波长检测不同杂质。故选择检测波长时,需对主成分及各已知杂质分别进行紫外光谱扫描,确定各自的最大吸收波长及其差异。
分析方法的开发不能一蹴而就,我们在方法开发的道路上砥砺前行、孜孜不倦,为提高药品质量的道路上贡献绵薄之力。

杭州澳亚生物技术股份有限公司(“澳亚生物”)是一家集产、学、研为一体,以无菌冻干粉针剂为主导产品的现代化制药企业。自1993成立以来,遵循“以人为本、质量为先”的可持续发展战略,澳亚生物已经逐渐从小型、简单的冻干生产,走向复杂、规模化的无菌生产模式,为客户提供全方位的无菌生产解决方案。

实事求是,以人为本
尊重流程,注重效率
