
2018年年关将至,289目录产品的一致性评价工作也要接近尾声,为了顺利在结点前通过一致性评价,挤入市场,各大药企都卯足了劲,加大马力去推动产品的一致性评价工作,通过药品上市目录集查询,药监局目前已公布了75个通过一致性评价的品规,成果只能算是差强人意。为了改变这种现状,企业除了继续加大药品一致性评价的推动力度外,还需要时刻关注药监局的政策变化,包括官方公布的参比制剂目录和可豁免或简化生物等效性实验的药品目录,这样可以少走弯路,达到事半功倍的效果。
目前为止,药审中心已经公布了十四批次共1069个药品品规的参比制剂目录和两批次共72个可减免生物等效性实验的药品目录,其中可豁免品种58个,可简化品种14个,药企可以在展开一致性评价工作前对照查阅,省去不必要的工作。这个可豁免或简化生物等效性实验的药品目录是根据《人体生物等效性豁免指导原则》的要求来确定的,而药监局这份指导原则部分是参照ICH M9编写的。近日,FDA公布了最新修订版的ICH M9指导原则草案,让我们来看看里面都有哪些内容吧。
两种含有相同活性成分且服用后在可接受的预定制范围内具有相同的生物利用度(药品吸收的速率和程度)的药品被认为具有生物等效性。范围是通过药品在体内的表现来确定的,例如安全性和有效性方面的相似性。在体内的生物等效性研究中,关键的药代动力学参数是AUC(药时曲线下面积)和Cmax(最大浓度),一般被用来评估药品吸收的速率和程度。
基于BCS(生物药剂学分类系统)的生物等效豁免方式可以减少体内的生物等效性研究,例如利用其他方法替代体内生物等效性研究。当药品在体内的表现可以通过体外数据来进行判断时,体内的生物等效性实验也许可以被豁免。药品BCS分类可以分为四类,Ⅰ类:高溶高渗、Ⅱ类:低溶高渗、Ⅲ类:高溶低渗、Ⅳ类:低溶低渗。基于BCS分类的生物等效性豁免只适用于速释口服固体制剂或者进入体循环的混悬制剂,治疗窗窄的药品不适用,但如果固定剂量的复方制剂中的各类药品成分满足要求也可获得生物等效性实验的豁免。
只有BCS分类为Ⅰ类和Ⅲ类药品才可获得生物等效性豁免,且成分需要和对照产品完全一致,任何盐基、酯类的改变,同分异构体或同分异构体的混合物都不适用,但是前药可以。
当药品的最大单人治疗规格能够在37±1℃和1.2-6.8的pH范围内,在250ml或更少的水相介质中完全溶解,被认为具有高溶解性,如果药品的最大规格无法满足该标准,但对照品可以,则需要提供额外的数据来证明药品是否可以适用基于BCS分类的生物等效性豁免。
01
溶出度
在进行体外溶出实验时,通常会使用摇瓶技术,并至少选择pH1.2、4.5、6.8三种缓冲溶液,来测定药品的溶解性。此外,药品在特定pH范围内的解离常数也应该进行评估,需要测量加入药品后的各种缓冲溶液的pH值和最后平衡的pH值确保溶出度的测定控制在特定的pH值范围内,在必要的时候需要对pH值进行调整。在1.2-6.8的pH范围内测量到的最小溶出度来判定药品的溶出度类别。
每种pH至少需要测量三次,来验证方法的稳定性,以符合药品的药典要求。此外,药品在溶剂中的稳定性也应该进行验证,以防药品不稳定降解度超过了10%而超出了溶解性的测量限度。溶解性不能被准确测量则无法判断药品的BCS分类,这种情况是不能适用基于BCS的生物等效性豁免的。除了实验数据,文献数据也可以用来证明支持药品的溶解性,因为审评文件中可能不能包括全部可以对药品进行判断的必要细节。
02
渗透性
药品渗透性的评估更倾向基于人体药代动力学研究的吸收程度,例如绝对生物利用度或质量平衡。
高渗性是指绝对生物利用度≥85%,当尿液中的母药含量或经过阶段1氧化和阶段2共轭代谢后的母药含量总量不少于口服剂量的85%时也可认为药品具有高渗性。一般来说,代谢产物仅指氧化和共轭代谢产物,降解和水解产物不包括在内,除非可以证明这类产物不是因为肠道的微生物作用产生的。未变化的药品不能用来判定吸收程度,除非有适当的数据证明,排出的母药来源于胆汁排泄、肠道分泌或者其他不稳定的代谢物,例如葡萄糖醛酸结合物、硫酸盐化合物、氮氧化物等,这些代谢物由于微生物的作用而重新变回母药。
公开发行的文献数据中有关体内研究的数据也可以被接受,因为审评文件中可能不能包括全部可以对药品进行判断的必要细节。渗透性也可以使用人结肠癌细胞应用经验证和标准化的体外方法进行判定,因为其可以模拟人体内的肠道药物吸收系统。人结肠癌细胞可以进行自发的形态学和生化性质的分化,利用顶端刷状缘、紧密的细胞间隔和几种活跃的运转蛋白来表达细胞极性,就像在小肠内部一样。由于可能缺少或没有转出蛋白(例如P-gp, BCRP, MRP2)和转入蛋白(例如PepT1, OATP2B1, 315 MCT1),使用人结肠癌细胞判定渗透性只限于被动运输的药品。
药品的渗透性按吸收程度分为0、低(<50%)、中等(50-84%)和高(>85%),用来实验的药品必须有足够的数量,每种渗透性检测建议不少于5个,并重复至少三次,这样才可以测到药品渗透性的可靠数据。在实验前后,人体结肠癌细胞需要通过对跨膜电阻抗(TEER)或其他指标进行检测,以确保其单层膜结构保持完整。此外,细胞单层膜结构的完整性可以使用已证明具有0渗透性的混合物进行判定。
如果通过质量平衡或体外人体结肠癌细胞研究来判定药物的高渗透性,药物在肠道系统中的稳定性文献数据也应该考虑进去,除非在尿液中有不少于85%的剂量被恢复成了原药。药品在肠道系统中的稳定性需要使用代偿及模拟胃和肠液或其他合适或相关的方法进行测定。药品溶液需要在37℃的体液环境下先孵化一段时间,例如,在胃液中孵化1小时,在肠液中孵化3小时,在此过程中降解程度超过10%的药品都被认为具有潜在的不稳定性。
方法验证
进行方法验证时需要选择足够多的不同渗透性的模型药物,将测试药品的均值、标准差和变异系数,胃肠系统体外的稳定性信息和数据支撑的被动运输机制考虑进去,建立高渗透性的内控标准,用来对测试药品的渗透性进行分类,目前用来验证渗透性的模型药物如下。
表一 渗透性分析方法验证用模型药物举例
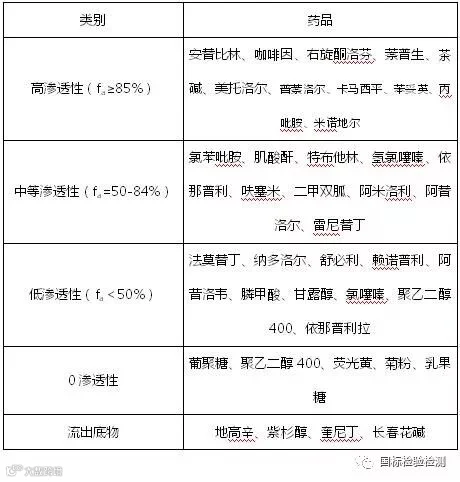
上文已经提到,人体结肠癌细胞判定药品的渗透性只限于被动运输药物,药物被动运输的机制可以通过对相关的临床剂量范围内的药代动力学数据进行推断。相应的,由于缺少主动蛋白转运机制,需要适合的分析系统来对被动运输进行分析,例如,选择药品在250ml溶剂中最大溶解剂量的0.01倍、0.1倍和1倍这几种浓度,通过流出系数计算转运方向来判定药品的体外渗透性。
流出系数= Papp基底外侧→顶端/Papp顶端→基底外侧
流出蛋白的功能表现通过对细胞膜两边流出转运底物的不对称性进行验证,流出转运底物一般选择不饱和的地高辛、长春花碱、罗丹明123溶液。渗透性研究中测试药品的浓度应该确定,经验证用于药品渗透性研究的人体结肠癌细胞方法的验证过程应包含所有情形,包括将中等和高渗透性的模型药物作为内控标准来证明方法的一致性,与测试药物使用同一溶液。选择的内控标准需要与测试药品相符合,不应该表现出任何特殊的理化或渗透作用。在渗透性检测时,内控标准和测试样品如果无法保持同样的细胞环境时,至少应该保持同一单层膜或同一平台的单层膜环境。内控标准的渗透率值在用不同方法检测时应该保持一致,即使是在方法验证期间。药品和内控标准在测试结束后的恢复率也应该进行评估,当恢复率小于80%时,质量平衡应该将残留在细胞膜上的药品计算在内。
选择合适的内控标准可以加快药品渗透性的检测过程,高渗透性的内控标准和中/高渗透性的分类界限十分接近,当渗透压值等于或大于所选的高渗透性内控标准时,测试药物被认为具有高渗透性。

没时间解释了,快长按左边二维码关注我们~