
上篇我们提到,当药品的最大单人治疗规格能够在37±1℃和1.2-6.8的pH范围内,在250ml或更少的水相介质中完全溶解,被认为具有高溶解性,如果药品的最大规格无法满足该标准,但对照品可以,则需要提供额外的数据来证明药品是否可以适用基于BCS分类的生物等效性豁免。以下是需要提供的数据:
03
基于BCS分类的药品生物等效性豁免的可行性支持
药品在满足某些标准的情况下,可以获得生物等效性实验的豁免,即属于BCSⅠ类和Ⅲ类,是全身作用的速释口服剂型,并与对照药物相比具有药学等效性。当测试药品的最大单次治疗剂量不能满足高溶出度标准,但对照药品可以,则需要额外的数据来支持生物等效性豁免,例如涵盖最大治疗剂量的剂量范围内的与剂量成比例的药代动力学数据,包括药时曲线下面积和最大浓度。
通过口腔或舌下吸收的药物不能适用基于BCS分类的生物等效性豁免,口腔分散片只有在不通过口腔和舌下吸收并且只用水送服的情况下才可以适用于基于BCS分类的生物等效性豁免。
为了保证药品可以满足以上条件,药品的辅料和体外溶出度都应该满足如下要求。
3.1
辅料
待测药品和对照药品内不同辅料对药品在体内的吸收影响应该进行评估,需要同时考虑药品成分的特性和辅料的作用,通过一种基于机制和风险的方法来确保变更的辅料不会影响药品主成分的吸收率和程度。
辅料可能会对溶出度、胃肠蠕动、转运时间和肠道通透性等产生影响,可能产生影响的辅料包括各种糖醇,如甘露醇,山梨醇和表面活性剂月桂醇硫酸酯钠盐,因此应该从多方面对辅料进行考量,包括用量、影响吸收作用的机制和药物主成分的吸收特性(速率、程度和吸收机制)。
可能会影响到待测和对照药品吸收的辅料在处方中的用量应该在开发过程中确定,并且变化保证在最低限。片剂包衣中和在特定药物影响界限范围内的少量辅料则不需过多的考虑。
根据定义,BCSⅠ类的药品都可以高度吸收,也没有溶出度或渗透性对吸收的限制。因此,该类药品辅料对药品吸收的影响风险低于其他药品。对于BCSⅠ类药品,我们一般考虑辅料对药品吸收速率和程度的潜在影响。例如,药品因为主动运输而具有高渗性,会影响药品主动运输的辅料就应该考虑进去;如果BCSⅠ类的药品吸收速率慢,则可以考虑加入促进吸收的辅料。
对于BCSⅠ类的药品,辅料的质量和数量可以在允许的范围内变动,但是会影响药物吸收的辅料要与对照药品具有相同的质量和相似的数量,变化不超过±10%。
表二 BCSⅠ类药品辅料变化情况举例
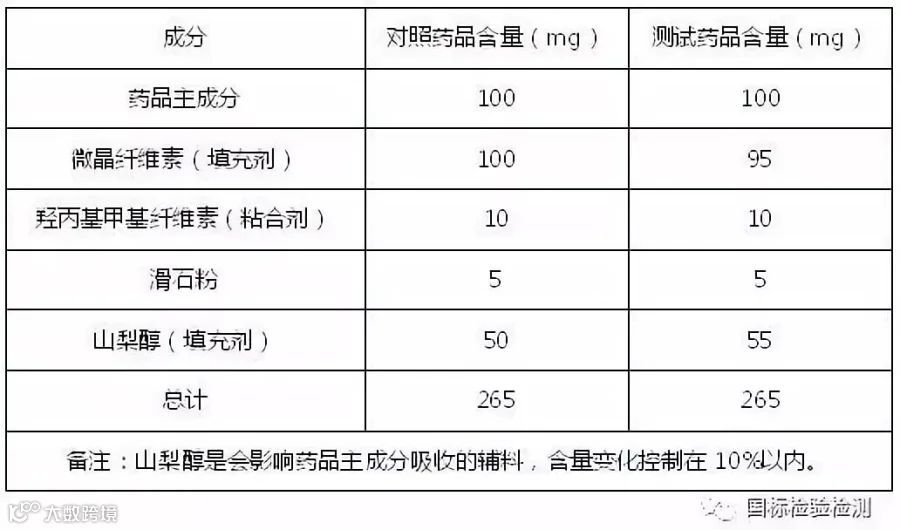
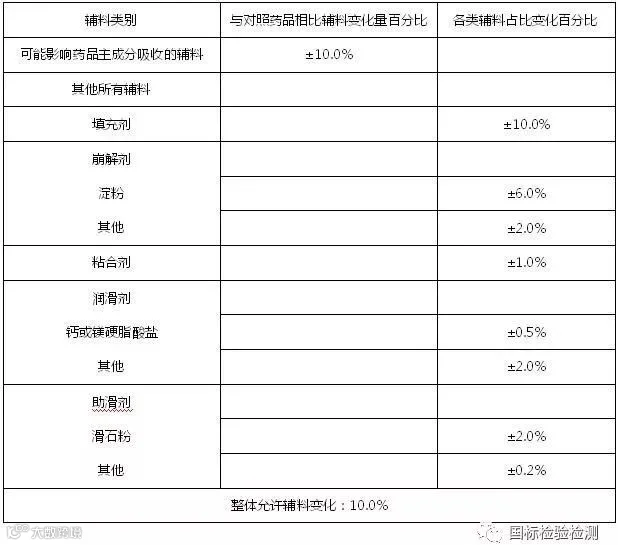
BCSⅢ类的药品更容易受辅料影响,这些药品渗透性不好只能在特定的部位吸收,因此相比BCSⅠ类药品来说,有更多的机制会使辅料对药物的吸收产生影响。对于BCSⅢ类的药品,所有的辅料都应该与对照药品质量一致、数量类似(除了包衣或胶囊壳用到的辅料),下面是BCSⅢ类药品允许的辅料变化范围。
表三 BCSⅢ类药品允许的辅料变化范围
表四 BCSⅢ类药品辅料变化情况举例
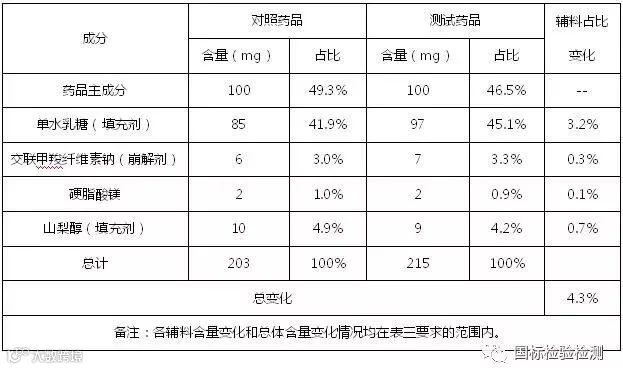
对于只含有BCSⅠ类药品的复方试剂,辅料变化标准按照BCSⅠ类药品执行;对于只含有BCSⅢ类或同时含有BCSⅠ类和BCSⅢ类的复方药品,辅料变化标准按照BCSⅢ类标准执行,对具有药学等效性的复方药物适用。
3.2
体外溶出度
在申请基于BCS分类的生物等效性豁免时,应该将与商业环境同一条件生产的一批测试药品和一批对照药品进行体外溶出度测试比较,测试药品的生产规模应该不少于一批药品注册规模的十分之一或10万个单位,两者取其高值。在临床研究阶段,小批量的规模也可以被接受,体外溶出度的比较试验应该使用药典设备和经验证的分析方法。
药品体外溶出度的比较试验应该满足如下特性:
(1)装置:桨或篮
(2)溶解介质体积:900ml或更少(按照QC测试的需求选择)
(3)溶解介质温度:37±1℃
(4)搅动:桨-50转/分钟,篮-100转/分钟
(5)至少各准备12个对照药品和测试药品
(6)缓冲剂:pH1.2、pH4.5和pH6.8,如果和上诉缓冲液不同,则需要对最小溶出度的pH缓冲液进行额外的研究,在某些情况下还需要用纯水作为额外的溶出介质
(7)不能使用有机溶剂,也不能添加表面活性剂
(8)在采集过程中应该对药品进行过滤
(9)对于具有明胶涂层的胶囊药物或片剂,可能会因为明胶的交联作用形成膜壳,可以加入适量的酶。
当50转/分钟的桨法有高变异性或悬浮性时,推荐使用100转/分钟的篮法。另外,经过确认后,在桨法中可以使用沉降篮来解决药品悬浮的问题。
BCSⅠ类药品满足基于生物等效性的豁免要求需要测试药品和对照药品在相同的特定外部条件下都表现出非常快速的溶出性(15分钟内平均溶出度超过85%)或快速的溶出性(30分钟内平均溶出度超过85%),并且曲线类似。万一一个产品表现出快速的溶出性同时对照药品表现出非常快速的溶出性,数据相似性可以通过下式判断:
f2 = 50 • log {[1 + (1/n)Σt=1 n (Rt - Tt) 2 ] -0.5 • 100}
式中f2是相似因子,n是取样点数,Rt是对照药品在时间t内的平均溶出度,Tt是测试样品在时间t内的平均溶出度,满足以下情况才可以使用相似因子进行判断:
(1)最少三个时间点(0点排除在外);
(2)测试和对照药品选择的时间点必须相同;
(3)每种药品每个时间点的均要取十二个药品溶出度的平均值;
(4)药物平均溶出度超过85%的取样点不超过一个;
(5)使用平均数据,早期时间点(10分钟以内)的变异系数(相对标准偏差)不超过20%,在其他时间点的变异系数不超过10%。
当f2值≥50时,则认为两条曲线相似。当测试和对照药品在15分钟内溶出度均超过85%,则不需要计算f2值,认为具有相似性。万一变异系数太高,f2值的计算结果则不准确不可靠,相关的相似性判断也不可信。
为了满足BCSⅢ类药品的生物等效性豁免资格,测试药品和对照药品在相同的特定外部条件下都要表现出非常快速的溶出度(15分钟内平均溶出度超过85%)。
对于复方制剂,溶出度曲线应该满足复方制剂中所有药品成分的标准,即含有BCSⅠ类药品的要满足BCSⅠ类药品的标准,含有BCSⅢ类要满足BCSⅢ类药品的标准,两种都含有的要满足两种药品的要求。
具有多种规格的药品,BCS分类的方式适用于其所有规格,一般来说测试样品和对照样品的溶出度曲线应该对所有规格进行比较。
04
文件
申请者应该提供测试和对照药品尽可能多且完善的关键质量属性信息,包括但不限于:多晶型和对映体纯度,任何有关药品生物利用度或生物等效性的信息,包括文件调研信息和衍生研究。所有研究草案包括的标准、质量确认和测试方法应该详细并按照最新的指导原则和法规进行过验证。
报告应该按照表格和图表展现所有数据和平均值,表格应该包括标准偏差和变异系数。
报告应该含有全部的辅料和他们的质量情况,如果有需要,测试药品和对照药品中辅料含量的不同也应该包括在内。
分析方法的完整描述,包括方法验证的线性、准确度和精密度。所有测试方法和介质的详细描述,包括测试和对照样品的批信息:剂量规格、批号、生产日期、批量、有效期等。溶出性报告应该包括对实验参数和分析方法完整详尽的描述,包括样品的溶出装置、通风设施、过滤设施、体积等。
另外,对于药品体外结肠癌细胞渗透性分析方法的完整信息在适用的时候也需要一并提供。

没时间解释了,快长按左边二维码关注我们~