简介
胺类化合物是氨分子中一个或多个氢原子被烃基取代后的有机化合物,化工业生产胺作为原料或终端产品,在医药化合物、农产品、精细化学品、染料和颜料等方面具有重要应用,因此胺的生产已经成为化学工业中增长最快的门类之一。 胺是药物活性分子的重要组成部分,它有助于提高药效,并优化所需的药物分子物理化学性质。根据Grand View Research, Inc.的研究报告,预计到2025年胺类化合物的市场规模预计将从2016年的144亿美元增长至293亿美元1。而在医药领域,据统计有超过90%的畅销药或新批准的小分子药物为胺类化合物或其衍生物,其中大多数为手性胺类化合物,并且在农药领域有大约30%的农药有效分子为手性胺类化合物2。
传统的胺类化合物的合成常采用催化氢化、金属还原法、硝基还原法、醛酮还原胺化、霍夫曼降解反应及亚胺的还原等方法3。采用化学方法合成胺类化合物常面临反应杂质多、路线长、环境污染大以及不对称胺类化合物合成中手性选择性差等缺陷。而生物催化的方法具有反应条件温和、杂质少、选择性高等优点,并且经过过去几十年的发展,目前通过生物催化方法合成胺类化合物的策略已受到普遍的重视,因其能够利用酶的高选择性、高催化活性的特点进行胺类化合物的合成。所涉及的催化酶类有转氨酶、亚胺还原酶、脂肪酶、海茵酶、氨基酸裂解酶、氨基酸脱氢酶、苏氨酸醛缩酶等,而利用转氨酶进行还原胺化反应制备胺类化合物的策略已被广泛采用2。
转氨酶是一类可逆催化氨基基团从氨基供体转移到氨基受体上以进行胺类化合物合成的酶。迄今为止发现的转氨酶均需要5-磷酸吡哆醛(PLP,维生素B6衍生物)作为辅因子。通常根据转氨酶所催化的底物与生成的产物的类型,将转氨酶分为α-转氨酶和ω-转氨酶,其中α-转氨酶催化的底物或产物中含有α-氨基酸结构,而ω-转氨酶催化α-氨基酸以外的底物4。其具体催化路线如图1所示,酶催化过程中PLP首先与酶共价结合以亚胺的形式(E-PLP)存在,催化结束辅因子释放氨,PLP回到最初状态,实现辅酶PLP的自动循环4,5。
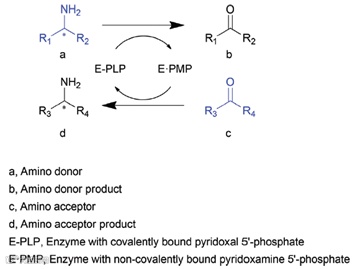
图1 转氨酶催化反应方程式5
具体地,转氨酶催化反应以乒乓反应机制进行(图2所示),由两个半反应组成:氨基供体的氧化脱氨和氨基受体的还原胺化。催化过程中PLP首先与催化氨基酸赖氨酸形成席夫碱,之后氨基底物取代赖氨酸,与PLP形成新的席夫碱,席夫碱发生水解形成相应的酮及酶PMP复合物。反应的后半部分,即还原胺化,是由E-PMP和氨基受体酮之间形成Michaelis complex引发的,PMP的脱氨基作用导致产物胺的形成和PLP的再生4,5。

图2 氨基供体的氧化脱氨机理4
在转氨酶作为催化剂制备胺类化合物的应用中,通常可分为两类,一类是对消旋胺类化合物进行动力学拆分获得高光学纯度的手性胺类化合物,另一类是以酮类化合物为底物,利用其高区域选择性和手性选择性进行的特点进行转氨反应制备胺类化合物。
(一)对消旋胺类化合物的动力学手性拆分(Kinetic resolution,KR)
动力学拆分是分离不同对映异构体的一种方法,适用于单一异构体的价值远高于消旋化合物的高手性纯度的化合物的制备。如图3所示,不同的对映异构体所处的吉布斯自由能级相同,进行相同反应的产物相同,但两者在进行相同反应过程中形成的过渡态的吉布斯自由能可能不同,这就使得对映异构体在进行相同反应时出现不一致的反应速率,反应较慢的对映异构体在剩余的未反应的底物中占优势,从而获得光学纯度高的手性化合物6。
在进行动力学拆分时,与不同对映异构体反应的手性试剂的选择极为关键,这将决定最终产品的光学纯度。在胺类化合物的化学拆分中,胺的高亲核性(与醇相比)会导致背景反应速率增加和损害反应的选择性(胺与大多数酰化试剂直接反应)。而酶具有选择性高,催化活性高的特点,是一种非常适合用于手性拆分的催化剂,其中转氨酶已被广泛应用于胺类化合物的手性拆分。转氨酶可选择性的催化其中一个胺类对映异构体与氨基受体酮进行转氨反应,生成相应的酮及副产物胺,而目标手性的胺则几乎不被消耗并且留在反应体系中成为优势产物,从而获得了高光学纯度的手性胺产品。

图3 动力学拆分的反应能量图与反应方案6
Ronald L. Hanson等人曾利用来源于巨大芽孢杆菌的S选择性转氨酶,以丙酮酸作为氨基受体,制备(R)-仲丁胺,具体技术路线如图4所示。其在50g/L消旋仲丁胺,25g/L S选择性转氨酶条件下过夜反应,最终获得了ee值为100%的24.3 g/L R-仲丁胺,反应收率达48.6%7。

图4 转氨酶拆分消旋仲丁胺制备R-仲丁胺7
(二)以酮为底物利用转氨酶的手性选择性不对称合成手性胺
利用转氨酶严格的手性选择性及催化高效性将酮底物直接转化为相应的手性胺类化合物是转氨酶合成胺类化合物领域中最为常见的应用。在(R)-α-苯乙胺的合成中,酶赛生物利用具有自主知识产权的BioEngine®酶定向进化平台开发了手性选择性高、催化活性高、稳定性好的工程化转氨酶用于(R)-α-苯乙胺的合成,反应以苯乙酮为底物,异丙胺为氨基供体进行转氨反应。该反应存在化学平衡,在水相反应体系中不易获得高浓度的产物积累。为克服化学平衡带来的不利影响,酶赛生物创新地使用固定化细胞全有机相体系进行反应,反应体系中仅含800g/L苯乙酮,2.0 M异丙胺,0.4 mM辅因子PLP,10 g/L水以及50g/L硅胶吸附的湿细胞,最终获得168g/L/d的(R)-α-苯乙胺的时空产率。同时通过反应装置(图5)的设计可轻松实现酶与反应液的分离,产品后处理更为便捷,另外未参与反应的苯乙酮及被硅胶吸附的酶可轻松实现回收套用,实现成本降低、绿色环保的生产工艺8。在(R)-1-(1-萘基)乙胺的合成中,酶赛生物同样以转氨酶为催化剂,以1-萘乙酮为反应底物,利用制备(R)-α-苯乙胺的相同的反应工艺,实现了(R)-1-(1-萘基)乙胺的产业化生产。

图5 全有机相体系转氨酶催化苯乙酮制备(R)-α-苯乙胺反应装置;
E-1: 硅胶固定化细胞; E-2:反应液储存罐; E-3: 蠕动泵8
(三)以酮为底物利用转氨酶的区域选择性和手性选择性不对称合成手性胺
在有机合成化合物过程中往往需要选择特定的官能团进行反应,根据国际理论与应用化学联合会(IUPAC)的定义,“区域选择性反应是其中一个键形成或断裂方向优先于所有其他可能方向发生的反应”9。使用化学催化剂往往较难控制反应的区域选择性,而酶作为催化剂则可实现较高的区域选择性。
胺类化合物中含有多个羰基或其它活泼基团的情况非常普遍,而在使用含多个活泼基团的前体作为底物来合成胺类化合物的过程中,羰基等活泼基团往往会同时发生反应,产生杂质。而酶具有较高的区域选择性,可选择性地只催化多羰基化合物中的一个羰基且只催化羰基进行转氨反应而不催化其它官能团,同时酶催化反应条件温和,可有效减少潜在的化学自发反应的发生,避免杂质的生成。
Stefan E. Payer等人利用转氨酶催化十五烷-2,5,8-三酮制备胺类化合物,所使用的转氨酶具有很高的区域选择性,其只选择性地催化底物2号位羰基进行转氨反应,而5号位与8号位的羰基则不被催化,该转氨酶不仅具备高区域选择性,也具备高手性选择性,可催化生成ee值为99%的手性胺产物。同时其采用多酶级联反应的策略,在反应体系中加入丙氨酸脱氢酶(AlaDH)使副产物丙酮酸转化为氨基供体丙氨酸,并利用甲酸脱氢酶(FDH)实现辅酶烟酰胺腺嘌呤二核苷酸(NAD)的循环,以此实现胺供体的循环及推动化学平衡10。

图6 转氨酶催化十五烷-2,5,8-三酮制备胺类化合物10
西他列汀磷酸盐是由美国默克公司开发,于2006年10月被美国FDA批准治疗II型糖尿病的药物,商品名为捷诺维(Januvia),西他列汀磷酸盐合成中的关键步骤是手性氨基中间体的构建。默克公司曾利用经定向进化改造的转氨酶催化剂催化西他列汀前体酮合成关键手性氨基中间体,在该反应中转氨酶展现出了高区域选择性及高手性选择性的优势,相较于化学法所获得的97% ee的产物,通过酶催化反应可获得99.95% ee的产物,反应单次底物投料量可达200g/L,反应收率达到92%11。

图7 A: 化学法合成西他列汀;B: 转氨酶催化西他列汀前体酮制备西他列汀11
(四)以酮为底物利用转氨酶的手性选择性进行动态动力学转氨反应(Dynamic kinetic transamination, DK-TA)
转氨酶催化过程中所使用的酮底物有时会具有手性结构(即含多个立体异构体),通常转氨酶催化生成的产物会要求严格的手性,包括非转氨反应产生的手性中心,也就是底物含有的手性中心。此时若直接采用单一构型的底物进行反应则底物的成本将会大大提高,若采用消旋的底物进行反应,则可有效的降低底物成本,但前提是消旋底物可100%地被利用,转化为手性胺产物。在保证产物高手性纯度的前提下,由于酶具有严格的手性选择性,通常消旋底物只能转化50%左右。而当底物存在一些特定结构时,如含有氢原子的手性碳原子与羰基相邻,则在某些条件下由于烯醇化的发生而使底物易于消旋化,实现不同底物异构体的互变,不被酶利用的底物在体系中发生消旋化,转化为可被酶利用的底物而逐渐被消耗,最终可实现消旋底物的100%转化。
乌布吉泮(Ubrogepant)是一种新型口服降钙素基因相关肽受体(CGRP) 拮抗剂,是一种偏头痛治疗药物,已于2019年被美国FDA批准上市,乌布吉泮含有2个关键前体,内酰胺(2)及螺酸(3)(图8),而内酰胺2的关键前体(10)(图9)则可通过动态动力学转氨反应制备。

图8 乌布吉泮合成关键前体12
Nobuyoshi Yasuda等人以消旋的酮为底物利用转氨酶进行动态动力学转氨反应合成乌布吉泮前体10,该乌布吉泮前体含有3个手性中心,其中5号位与6号位的高手性纯度通过转氨酶的手性选择性控制。Nobuyoshi Yasuda等人设计了高温(55℃)、强碱性(pH10.5)及高有机溶剂浓度(50% DMSO)的反应条件以实现底物的快速消旋化,同时为使酶能够在如此严苛的反应条件下保持催化活性及良好的手性选择性,Nobuyoshi Yasuda等对酶进行针对性的定向进化以提高酶的催化活性,催化稳定性及手性选择性,最终在50g/L底物条件下反应24h,反应收率可达92%,产品dr值可达60:1,产品提纯收率可达80%12。

图9 转氨酶催化反应制备乌布吉泮关键前体12
Dominik Koszelewski等人以消旋的4-氧代-3-苯基丁酸乙酯(图10,rac-3)为底物进行胺类化合物的合成,该转氨酶催化醛基进行转氨反应,故转氨反应并不会产生新的手性中心,转氨反应产物的手性中心由底物引入,且要求产品的手性构型为R构型。Dominik Koszelewski等人使用的转氨酶对底物具有良好的选择性,可选择性地催化R构型底物进行反应从而获得高纯度的R构型产物。由于与底物手性碳原子与羰基及苯环相邻,与其相连的氢较为活泼,易发生酮式-烯醇式互变,使底物的消旋化得以发生,底物可被转氨酶100%的转化。该反应也同样引入级联反应,在反应体系中加入LDH消耗反应生成的丙酮酸以推动化学平衡13。

图10 转氨酶催化消旋4-氧代-3-苯基丁酸乙酯制备胺类产品13
(五)转氨酶在级联反应中的应用
除利用单一的转氨酶制备手性胺类化合物之外,转氨酶常与其它酶类如酮还原酶、氧化酶、醛酸酶等进行联用,制备多手性中心的高手性纯度的胺类化合物。
(2S,3R)-2-氨基-1,3,4-丁三醇(ABT)是合成HIV蛋白酶抑制剂奈非那韦的前体,并且作为合成解毒素的中间体,可降低稻瘟病抗生素治疗的毒性。MBA可以由简单的非手性起始材料通过偶联转酮醇酶和转氨酶催化制备。Pia Gruber等人以羟基丙酮酸(HP)为起始物料进行ABT的合成,整个合成由两部分组成:首先起始原料羟基丙酮酸与乙醇醛在转酮醇酶的催化下生成中间产物L-赤藓酮糖,之后L-赤藓酮糖在转氨酶的催化下生成产物ABT。

图11 转酮醇酶和转氨酶的级联反应制备ABT14
针对该级联反应,Pia Gruber等人开发了相应的反应装置,转酮醇酶和转氨酶的催化反应在不同的反应模块中进行,同时增加了萃取模块,使反应结束后的ABT得以从体系中分离14。
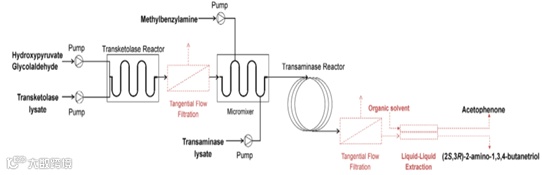
图12 ABT制备反应装置14
转氨反应通常具有严重的化学平衡问题,为推动化学平衡常采用添加过量氨基供体,移除副产物等方式,而采用级联反应移除副产物是较为常用且有效的推动化学平衡的方式。
草铵膦(PPT)是一种低毒、非选择性除草剂,广泛用于转基因作物。但目前商用的PPT是含有约50% D-PPT的外消旋混合物,无除草活性,对环境有害。因此,有必要研究一条合成光学纯L-PPT的可行路线。Feng Cheng等人开发了一种单转氨酶催化的级联反应制备高手性纯度的L-PPT,其以2-氧代-4-[(羟基)(甲基)膦酰基]丁酸(PPO)为起始原料,谷氨酸作为氨基供体,经转氨酶催化生成产物L-PPT及副产物α-酮戊二酸15。该转氨酶也具有催化α-酮戊二酸与L-天冬氨酸进行转氨反应的活性,为移除副产物α-酮戊二酸,Feng Cheng等人又向反应体系中加入L-天冬氨酸,使α-酮戊二酸转化成胺供体谷氨酸,并生成副产物草酰乙酸。草酰乙酸不稳定易脱酸生成丙酮酸,而该转氨酶对催化丙酮酸活性极低,几乎不会对丙酮酸发生转氨反应,从而实现副产物的移除,进而推动化学平衡。采用该反应策略,Feng Cheng等人在大规模反应中,使用10 kg表达转氨酶的大肠杆菌细胞作为催化剂,在80 kg PPO的投料量下反应24小时,获得ee > 99%的PPT产物70 kg。
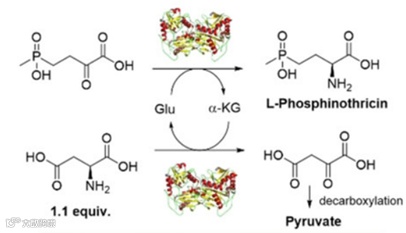
图13 单转氨酶级联反应制备L-PPT15
结语
转氨酶作为制备胺类化合物的生物催化剂,经过过去几十年的发展已逐渐成熟,结合酶定向进化技术,转氨酶在合成胺类化合物的应用场景也会越来越多样。尤其在强调“碳中和”环保发展理念的今天,生物催化法制备化合物的优势也将逐渐显现。
撰稿 | 胡虎 戴怡昕 蔡宝琴 周雪斓
参考文献
[1] Dr.Nadim S.Shaikh. Sustainable Amine Synthesis: Iron Catalyzed Reactions of Hydrosilanes with Imines, Amides, Nitroarenes and Nitriles[J]. ChemistrySelect,2019,4(22).
[2] Wu Shuke,Snajdrova Radka,Moore Jeffrey C,Baldenius Kai,Bornscheuer Uwe. Biocatalysis: Enzymatic Synthesis for Industrial Applications.[J].Angewandte Chemie(International ed.in English),2020,60(1).
[3]何永富,李荣疆.胺的合成反应综述[J].合成化学研究,2016,4(2):11-18.
[4] Brundiek H,M Höhne. Transaminases–A Biosynthetic Route for Chiral Amines[M].Wiley‐VCH Verlag GmbH & Co.KGaA,2016.
[5] Guo Fei, Berglund Per. Transaminase biocatalysis: optimization and application[J].Green Chemistry,2017,19(2).
[6] Robert E. Gawley. Do the Terms“%ee” and “% de” Make Sense as Expressions of Stereoisomer Composition or Stereoselectivity?[J].The Journal of Organic Chemistry,2008.
[7] Hanson R, Davis B, Chen Y, et al. Preparation of(R)-Amines from Racemic Amines with an (S)-Amine Transaminase from Bacillus megaterium[J].Advanced Synthesis& Catalysis,2008.
[8] Cai B,J Wang, Hu H ,et al. Transaminase Engineering and Process Development for a Whole-Cell Neat Organic Process to Produce ( R )-α-Phenylethylamine[J]. Org. Process Res. Dev.2022,26,7,2004–2012.
[9] Riva S. Regioselectivity of hydrolases in organic media[J]. Springer Netherlands,1996.
[10] Stefan E. Payer,Joerg H. Schrittwieser,Barbara Grischek,Robert C. Simon,Wolfgang Kroutil. Regio‐ and Stereoselective Biocatalytic Monoamination of a Triketone Enables Asymmetric Synthesis of Both Enantiomers of the Pyrrolizidine Alkaloid Xenovenine Employing Transaminases[J]. Advanced Synthesis & Catalysis,2016,358(3).
[11] Savile Christopher K.,Janey Jacob M.,Mundorff Emily C.,Moore Jeffrey C.,Tam Sarena,Jarvis William R.,Colbeck Jeffrey C.,Krebber Anke,Fleitz Fred J.,Brands Jos,Devine Paul N.,Huisman Gjalt W.,Hughes Gregory J.. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture[J]. Science,2010,329(5989).
[12] Yasuda Nobuyoshi,Cleator Ed,Kosjek Birgit,Yin Jianguo,Xiang Bangping,Chen Frank,Kuo Shen Chun,Belyk Kevin,Mullens Peter R.,Goodyear Adrian,Edwards John S.,Bishop Brian,Ceglia Scott,Belardi Justin,Tan Lushi,Song Zhiguo J.,DiMichele Lisa,Reamer Robert,Cabirol Fabien L.,Tang Weng Lin,Liu Guiquan. Practical Asymmetric Synthesis of a Calcitonin Gene-Related Peptide (CGRP) Receptor Antagonist Ubrogepant[J]. Organic Process Research & Development,2017,21(11).
[13] Koszelewski Dominik,Clay Dorina,Faber Kurt,Kroutil Wolfgang. Synthesis of 4-phenylpyrrolidin-2-one via dynamic kinetic resolution catalyzed by ω-transaminases[J]. Journal of Molecular Catalysis. B, Enzymatic,2009,60(3).
[14] Gruber P , Carvalho F , Marques M , et al. Enzymatic synthesis of chiral amino-alcohols by coupling transketolase and transaminase-catalyzed reactions in a cascading continuous-flow microreactor system[J]. Biotechnology and Bioengineering, 2017.
[15] Cheng Feng,Li Ju Mou,Zhou Shi Peng,Liu Qi,Jin Li Qun,Xue Ya Ping,Zheng Yu Guo. A single-transaminase-catalyzed biocatalytic cascade for efficiently asymmetric synthesis of L-phosphinothricin.[J]. Chembiochem : a European journal of chemical biology,2020,22(2).
