

来源:公众号npj计算材料学

基于经验力场的经典分子动力学 (MD) 已成为一种在原子尺度上预测材料性质和行为的强大技术。与基于密度泛函理论 (DFT) 的从头计算方法相比,使用原子间相互作用的经验模型,可以显着提高计算速度和可扩展性。MD 模拟的核心组件是描述原子间相互作用的原子间势 (IP),IP 的选择会显著影响相关材料性质预测的结果,从而影响结果分析。此前已有研究表明,IP对参数的微小变化非常敏感。此外,由于这种IP具有严格的物理形式,它们无法在不同的材料系统中进行推广。最近,研究者们通过机器学习(ML)的方法来开发IP。利用机器学习开发IP最热门的方法包括线性回归、核回归模型和神经网络。这些模型对 IP 的物理形式的假设较少,且很容易推广到不同的材料系统中。然而,这些方法存在许多问题。例如,核回归模型需要大量的计算资源来进行数据输入和参数估计,造成了严重的计算和内存瓶颈。

Fig. 1 Prediction accuracy of potential based on random features.
来自多伦多大学的Gurjot Dhaliwal等人提出了一种可以高效模拟原子间相互作用(如能量和力)的计算方法。他们提出的广义线性模型使用随机特征的线性组合来近似模拟能量和力,并基于此解决线性最小二乘问题实现快速参数估计。该方法分别用随机傅里叶特征和随机特征图来近似静止核和非静止核,利用机器学习有效地从DFT模拟数据集学习IP。研究发现,基于ML开发的IP具有通用的函数形式,可以容易地推广应用于不同类别的复杂材料。利用这种方法的MD模拟获得的能量和力,与DFT计算值完美吻合。更重要的是,该模型的低维参数空间和线性函数形式极大地提高了计算效率,减低了运行成本,计算速度比 DFT 更快,且允许更快的超参数搜索和优化。该文近期发布于npj Computational Materials 8: 7 (2022) 。

Fig. 2 Comparison of phonon properties.
Editorial Summary
Machine learned interatomic potentials using random features
Classical molecular dynamics (MD) based on empirical force fields has emerged as a powerful technique for predicting material properties and behaviours at the atomistic scale. This is because the use of empirical models of interatomic interactions enables significantly faster and scalable computations compared to ab initio methods such as density functional theory (DFT). The central component of any MD simulation is the Interatomic Potential (IP) function that determines interatomic interactions. The choice of IP can significantly alter the property predictions, thus affecting the derived insights. It has been shown that IPs can be sensitive to small changes in their parameters. Furthermore, due to their rigid physically motivated functional forms, they cannot be generalized across different material systems. In recent years, machine learning (ML) tools are widely used to develop IPs. The most popular ML approaches for IP development include linear regression, kernel regression models, and neural networks. These models make fewer assumptions on the functional form of the IP and can be easily generalized to different material systems. However, problems still exist. For instance, kernel regression model requires a lot of computing resources for data input and parameter estimation, resulting in significant computational and memory bottleneck.
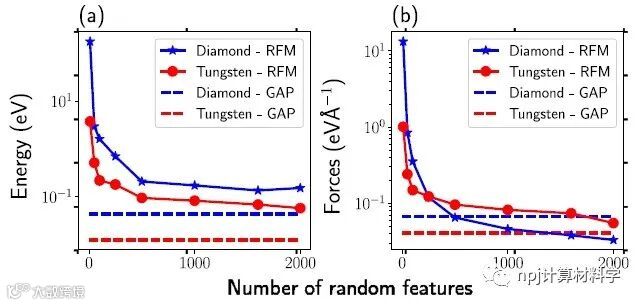
A team led by Prof. Gurjot Dhaliwal from the Department of Mechanical and Industrial Engineering, University of Toronto, presents a method to model interatomic interactions such as energy and forces in a computationally efficient way. The proposed model approximates the energy/forces using a linear combination of random features, thereby enabling fast parameter estimation by solving a linear least-squares problem. Random Fourier features and Random features map corresponding to stationary and non-stationary kernels are used to efficiently learn IPs from DFT simulation datasets. It is found that ML-based IPs can outperform classical empirical IP, by using a generic functional form, with DFT level accuracy for energy/force prediction. The functional form can be easily extended to model complex material interactions that involve different material classes. Meanwhile, MD simulations performed using the IPs based on random features provided better energy/force fittings. More importantly, low parametric space and linear functional form enable the model to be computationally faster than DFT and allow faster hyper parameter search and optimization. This article was recently published in npj Computational Materials 8: 7 (2022) .
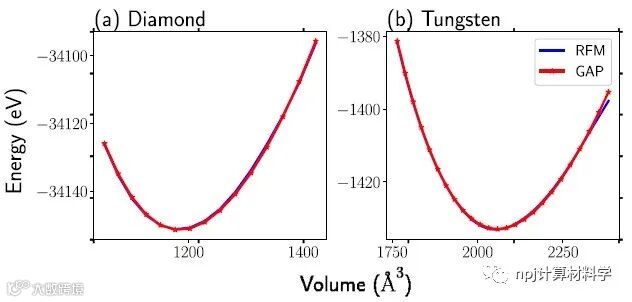
Fig. 4 Fitting equation of states for diamond and tungsten data.
原文Abstract及其翻译
Machine learned interatomic potentials using random features (使用随机特征的机器学习原子间势)
Gurjot Dhaliwal, Prasanth B. Nair & Chandra Veer Singh
Abstract We present a method to model interatomic interactions such as energy and forces in a computationally efficient way. The proposed model approximates the energy/forces using a linear combination of random features, thereby enabling fast parameter estimation by solving a linear least-squares problem. We discuss how random features based on stationary and non-stationary kernels can be used for energy approximation and provide results for three classes of materials, namely two-dimensional materials, metals and semiconductors. Force and energy predictions made using the proposed method are in close agreement with density functional theory calculations, with training time that is 96% lower than standard kernel models. Molecular Dynamics calculations using random features based interatomic potentials are shown to agree well with experimental and density functional theory values. Phonon frequencies as computed by random features based interatomic potentials are within 0.1% of the density functional theory results. Furthermore, the proposed random features-based potential addresses scalability issues encountered in this class of machine learning problems.
摘要 我们提出了一种可以高效模拟原子间相互作用(如能量和力)的计算方法。该提出的模型使用随机特征的线性组合来近似模拟能量和力,基于此,解决线性最小二乘问题来实现快速的参数估计。我们讨论了如何用基于静止和非静止核的随机特征来进行能量近似,并将此应用于二维材料、金属和半导体,计算了这三类材料的能量近似值。使用上述方法,计算获得的能量和力的预测值,与密度泛函理论结果高度一致,并且模拟过程的训练时间比标准核模型低96%。我们进行了基于该随机特征原子间势的分子动力学计算,其结果与实验值和密度泛函理论值也高度吻合。基于随机特征的原子间势计算的声子频率,与密度泛函理论结果的差值不到 0.1%。此外,基于随机特征的原子间势方法解决了机器学习中的遇到的非扩展性问题。
免责声明:本文旨在传递更多科研资讯及分享,所有其他媒、网来源均注明出处,如涉及版权问题,请作者第一时间后台联系,我们将协调进行处理,所有来稿文责自负,两江仅作分享平台。转载请注明出处,如原创内容转载需授权,请联系下方微信号。
