
劲爆消息! 《纳米结构材料》所有章节内容PDF文件均可下载!(点击进入>>)

图1. 五种典型的异质结系统.
要点:根据两个半导体相对带的位置,可以分为三种类型的异质结: I, II; 根据电子转移的方式不同,可以分为p-n型和两种Z型异质结。
表1. 论文中出现的Table 1 Typical nano-heterophase junctions of photocatalysts.

https://mp.weixin.qq.com/s/zp_KTVh2CV87r-1d9vyg3Q

https://mp.weixin.qq.com/s/NFIt48ZItdP9dNiMNZ-Ydw
【一】小白也能看懂的PN结形成过程
冷知然 欲知其然 2022年03月03日 21:34

1 原子结构与结构示意图
1.1 原子基本结构
在高中化学中我们知道,原子是指化学反应不可再分的基本微粒。原子在化学反应中不可分割,但在物理状态中可以分割。物质是由原子构成的,原子基本组成结构如下图所示 。


其中:
-
质子带正电,核外电子带负电,质子数=核外电子数,故原子呈电中性
-
中子不带电
1.2 原子结构示意图
从上述可知,原子外围有许多带负电的电子,且这些电子并不是在同一轨道上运动的,而是分成排列的,如下图所示。

具有以下特点:
-
第一层最多2个电子,第二层最多8个电子,当电子层超过三层时,倒数第二层不超过18个电子;当电子层超过四层时,倒数第三层最多不超过32个电子,最外层不超过8个电子。
-
最外层8个电子的结构叫做稳定结构(特殊的是稀有气体中的氦是最外层2个电子)
-
每层最多排2n²个电子。(n表示层数)
-
金属原子:最外层电子数<4 易失电子
-
非金属原子:最外层电子数≥4 容易得到电子
-
半导体原子:最外层电子数=4 不易失去电子也不易得到电子(如上图为硅原子结构示意图,最外层有四个电子)
-
电子所在层数越高能量越大:站得越高摔得越痛(比喻),也可以理解为:质子带正电,电子带负电,异性相吸,离得越远,需要把它俩拉开的能量越大。
2 本征半导体
-
定义:完全纯净的、结构完整的半导体晶体,称为本征半导体。相对的,不纯净的半导体称为杂质半导体。 -
半导体晶体结构:在半导体晶体中(如硅和锗晶体),每个原子与其相邻的原子之间共用一对价电子达到8电子稳定结构,原子之间形成共价键,如下图所示。共价键有很强的结合力,使原子规则排列,形成晶体。 -
晶体特点:共价键中的两个电子被紧紧束缚在共价键中,称为束缚电子,常温下束缚电子很难脱离共价键成为自由电子,因此本征半导体中的自由电子很少,所以本征半导体的导电能力很弱。

3 杂质半导体
-
定义:不纯净的半导体称为杂质半导体,掺入某些微量的杂质。 -
作用:提高导电能力。 -
分类:

记忆:
-
N:负面的,因为多了一个带负电的电子 -
P:正面的,因为产生了一个带正电的空穴
4.1 N型半导体
在本征半导体(如硅原子)中掺入少量的5电子原子(如磷原子),该原子代替了4电子的位置,此时磷原子周围就有9个电子,原本只要8个电子就能达到稳定,而现在多出来一个电子,这个电子受原子核的束缚很小,很容易挣脱束缚变成自由电子,在晶体中到处游动,这就增加了半导体的导电性能。

4.2 P型半导体
在本征半导体(如硅原子)中掺入少量的3电子原子(如硼原子),该原子代替了4电子的位置,此时磷原子周围只有7个电子,原本要8个电子才能达到稳定,而现在少了一个电子(空出了一个位置——称为空穴),结构处于非稳定状态,原子核会吸引晶体中的自由电子填补空位以达到稳定状态,这样使稳定的半导体产生了电子移动,增加了半导体的导电性能。

4.3 杂质半导体的示意表示法
-
P型半导体:多空穴(带正电) -
N型半导体:多电子(带负电)

4 PN结
4.1 PN结的形成
PN结是不是直接将P型半导体和N型半导体黏合在一起就变成PN结了呢?
非也,非也。
PN结的形成是在同一片半导体基片上,分别制造P型半导体和N型半导体,经过载流子的扩散,在它们的交界面处就形成了PN结,如下图所示。

4.2 扩散运动(浓度差形成——内因)
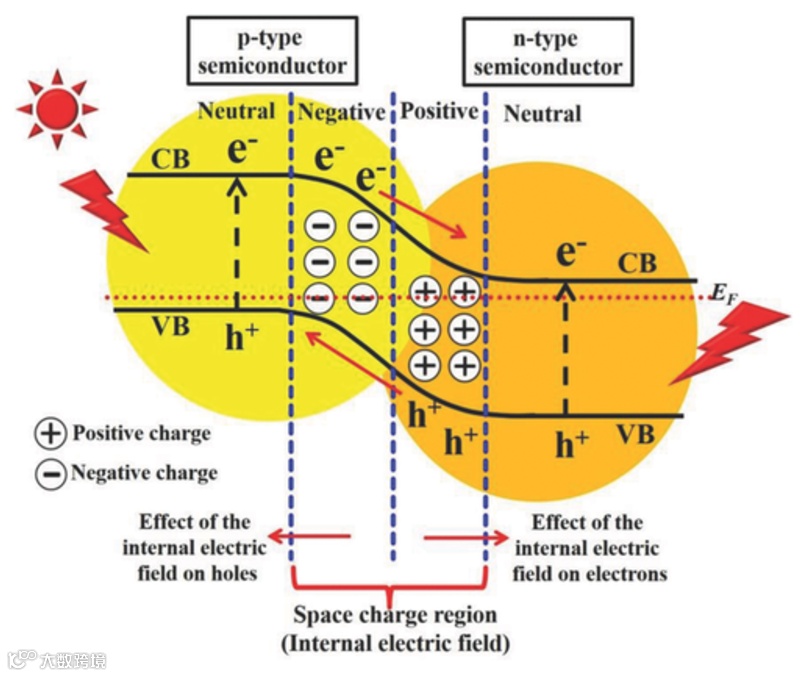
物质总是从浓度高的地方向浓度低的地方运动,因浓度差而产生的运动称为扩散运动。如下图所示,由于两个杂质半导体制成一体时,交界处两种载流子的浓度差很大,自由电子从N型半导体向P型半导体移动,相对的,空穴从P型半导体向N型半导体移动,当两种载流子接触时就会复合以能量的形式流失,剩下不能移动的正离子和负离子,由于正负离子的存在形成了电场,即内建电场(也称空间电荷区)。

4.2 漂移运动(电场力作用——外因)
在电场力的作用下,载流子的运动称为漂移运动。
4.2.1 正向偏置
当给P型半导体接正极,N型半导体接负极时,正极不断向P型半导体注入正电子(空穴),负极不断给N型半导体注入负电子(自由电子),增加两种载流子在交界处的浓度差,使两种载流子复合速率加快,进而使内建电场(空间电荷区)变小。

4.2.2 反向偏置
当给P型半导体接负极,N型半导体接正极时,正极不断向P型半导体注入负电子(自由电子),P型半导体中的空穴与注入的自由电子复合,同理,正极不断给N型半导体注入正电子(空穴),N型半导体中的自由电子与注入的空穴复合,减少两种载流子在交界处的浓度差,使两种载流子复合速率降低,进而使内建电场(空间电荷区,也称耗尽层)变大。

冷静认真多看几遍,每一遍都有不一样的收获!
参考文献
-
[1]:夸克(物理名词)_百度百科 (baidu.com)
【二】异质结类型的介绍:传统和新型异质结
材料er 2024年11月23日 09:39
为了有效分离半导体中光生成的电子-空穴对,人们提出了各种策略,例如通过掺杂、 金属负载、或引入异质结。在这些策略中,光催化剂中的异质结工程因其在空间上分离电子-空穴对的可行性和有效性,已被证明是制备先进光催化剂的最有前途的方法之一。
传统的三种异质结
跨隙型(I 型)、交错隙型(II 型)和断隙型(III 型)。
对于 I 型异质结光催化剂,半导体 A 的导带 (CB) 和价带 (VB) 分别高于和低于半导体 B 的相应带 。由于电子和空穴都聚集在同一半导体上,I 型异质结光催化剂的电子-空穴对无法有效分离。此外,氧化还原反应发生在氧化还原电位较低的半导体上,从而大大降低了异质结光催化剂的氧化还原能力。
II 型异质结光催化剂,半导体 A 的 CB 和 VB 位置高于半导体 B 的相应位置。因此,在光照射下,光生电子将转移到半导体 B,而光生空穴将迁移到半导体 A,从而导致电子-空穴对的空间分离。与 I 型异质结类似,II 型异质结光催化剂的氧化还原能力也会降低,因为还原反应和氧化反应分别发生在还原电位较低的半导体 B 和氧化电位较低的半导体 A 上。
III 型异质结光催化剂的结构与 II 型异质结光催化剂相似,只是交错间隙变得非常大,以至于带隙无法重叠。因此,III 型异质结无法实现两种半导体之间的电子-空穴迁移和分离,不适合用于增强电子-空穴对的分离。
在上述传统异质结中,II 型异质结显然是用于提高光催化活性最有效的传统异质结,因为它具有适合电子-空穴对空间分离的结构。常见II 型异质结:TiO2/g-C3N4、 BiVO4/WO3、g-C3N4-WO3 等,以提高光催化活性。一般来说,II 型异质结光催化剂具有良好的电子-空穴分离效率、较宽的光吸收范围和较快的传质速度。

p-n 异质结
p-n 异质结光催化剂的概念就是通过提供额外的电场,加速电子-空穴在异质结上的迁移,从而提高光催化性能。在光照射之前,p-n 界面附近 n 型半导体上的电子往往会扩散到 p 型半导体中,留下带正电的物质。同时,p-n 界面附近的 p 型半导体上的空穴会扩散到 n 型半导体中,留下带负电的物质。电子-空穴扩散将持续到系统达到费米级平衡为止。因此,靠近 p-n界面的区域会带电,形成一个 "带电 "空间或所谓的内部电场。当 p 型和 n 型半导体受到能量等于或高于其带隙值的入射光照射时,p 型和 n 型半导体都会被激发,产生电子-空穴对。在内部电场的影响下,p 型半导体和 n 型半导体中光生成的电子和空穴会分别迁移到 n 型半导体的 CB 和 p 型半导体的 VB,从而导致电子-空穴对的空间分离。值得注意的是,这种电子-空穴分离过程在热力学上也是可行的,因为在 p-n 异质结光催化剂中,p 型半导体的 CB 和 VB 位置通常高于 n 型半导体。
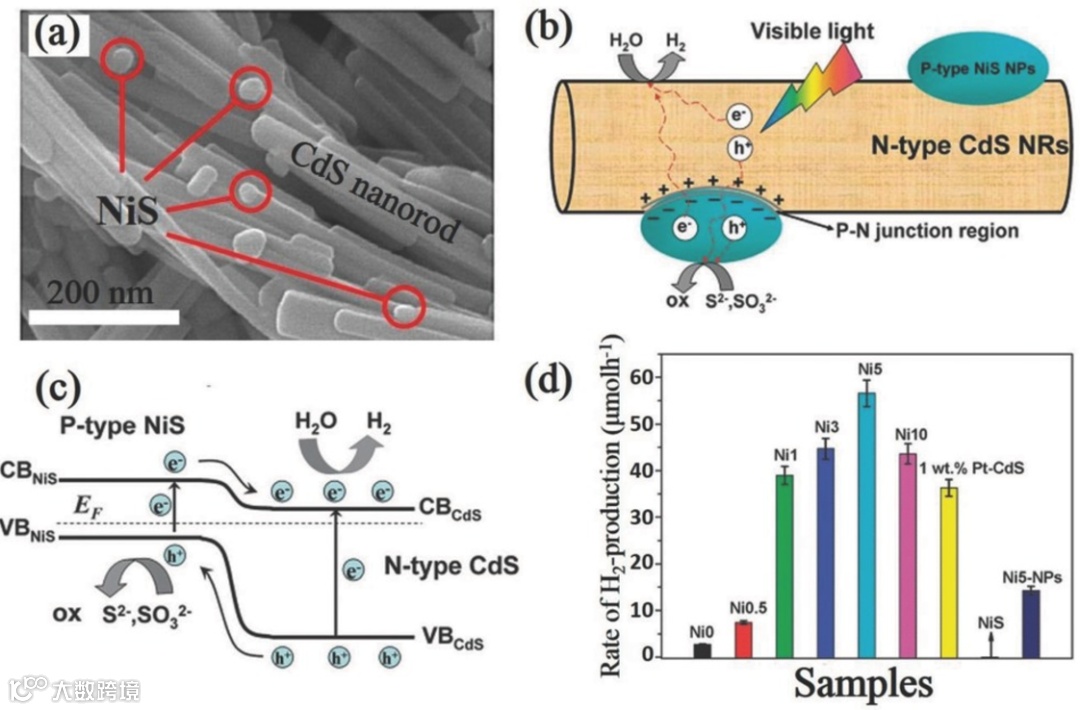
NiS/CdS p–n heterojunction:Phys. Chem. Chem. Phys. 2013, 15, 12088.
表面异质结
表面异质结是在单个半导体晶面上观察到的独特的电子-空穴分离现象。 众所周知,单个半导体的不同晶面可能具有不同的带状结构、 由于异质结是由两种具有不同带状结构的半导体材料组合而成,因此有可能在单个半导体的两个晶面之间形成异质结,即表面异质结。

J. Am. Chem. Soc. 2014, 136, 8839.
Z 型异质结
在光照射下,PS II 的 VB 上的电子首先被激发到 CB 上,在 VB 上留下空穴。然后,PS II 上的光生电子迁移到 PS I 的 VB 上,并进一步被激发到 PS I 的 CB 上。因此,光生空穴和电子分别积聚在氧化电位较高的 PS II 和还原电位较高的 PS I 中,从而实现了电子-空穴的空间分离和氧化还原电位的优化。
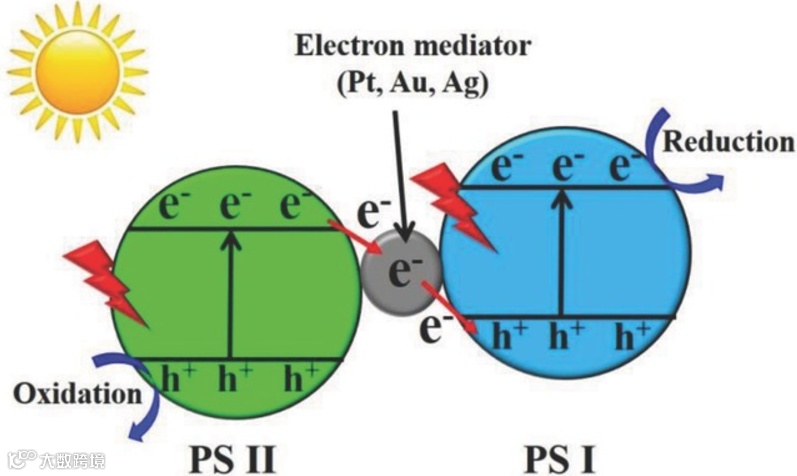

Phys. Chem. Chem. Phys. 2013, 15, 16883.
参考文献:Adv. Mater. 2017, 29, 1601694
【三】S型异质结和Z型异质结: 基本定义和案例分析!
【1】“纳米复合材料”和“纳米异质结”的异同点
(1)纳米复合材料:是由两种或两种以上物理化学性质不同的物质组合而成的一种多相固体材料,其中至少有一种是纳米尺度的微粒、纤维或薄膜。这些不同形态的纳米材料统称为分散相,“包裹”它们的材料称为基体。分散相形态不一,材料种类众多,但都满足一个最基本的要求:某一维度的尺寸在纳米尺度,由此带来的纳米效应是纳米复合材料具有奇异物理和化学特性的根本原因。纳米效应包括表面效应、尺寸效应和宏观量子隧道效应【1】。
(2)异质结:通常由两种不同性质的半导体单晶薄层构成,但在结合面须保持晶格的连续性,因而这两种材料至少要在结合面上具有相近的晶体结构。用两种单晶材料构成异质结必须满足晶格匹配和热匹配的要求【2】。异质结(heterojunction)是指两种不同的半导体材料的界面,它们的原子结构和电子能带结构不同。在异质结中,由于半导体材料的不同,界面处的电子能带结构会发生变化,这会导致界面处产生一种特殊的电场。异质结在半导体器件中具有重要的作用,因为它们可以用来提高器件的性能。例如,在太阳能电池中,异质结可以用来提高电池的转换效率;在发光二极管(LED)中,异质结可以用来提高发光亮度。此外,异质结还可以用来制造其他的半导体器件,例如晶体管、集成电路等。总的来说,异质结在半导体器件中具有重要的作用,因为它们可以提高器件的性能,并且可以用来制造许多不同的半导体器件。根据能带之间的相互关系,传统的异质结分为三种,跨立型 (straddling gap),错开型 (staggered gap)和破隙型 (broken gap),如下图【3】。

图1 传统的三种异质结光催化剂
在光催化的研究过程中,光生电子空穴复合严重以及单个光催化剂氧化还原能力低导致总的光催化反应效率很低的问题总是被人诟病。为了缓解这一问题,各种异质结技术被发展出来,目前被广泛认同的异质结技术主要包括:Type-II异质结和Z型异质结。最近,武汉理工大学余家国教授在Chem上发表了题为“S-Scheme Heterojunction Photocatalyst”的观点文章,详细阐述了所谓S型异质结,其在电荷转移机理方面同其他两种异质结的不同,以及局限性和未来的发展方向。在对传统异质结理解的基础上,一种新的S型异质结的概念被提出。该概念首次被余家国教授课题组于2019年提出。[3] 其全称叫做step-scheme heterojunction。这种异质结主要由功函数较小、费米能级较高的还原型半导体光催化剂(RP)和功函数较大、费米能级较低的氧化型半导体光催化剂(OP)通过错开型方式构建而成,可以有效实现强氧化还原能力电子空穴对的分离【4】。

图2 S型异质结中电荷转移过程
问题和解答
问题1:有没有异质结合成成功的大佬啊请教一些问题:两种复合材料是属于两种材料各自合成后吸附在一起的吗?还是一种在另一种生长上去的?
答案:通常形成异质结的条件是:两种半导体有相似的晶体结构、相近的原子间距和热膨胀系数。利用界面合金、外延生长、真空淀积等技术,都可以制造异质结。异质结是由两种不同的半导体材料相接触所形成的界面区域。按照两种材料的导电类型不同,异质结可分为同型异质结(p-p结或n-n结)和异型异质(p-n或p-n)结,多层异质结称为异质结构。通常形成异质的条件是:两种半导体有相似的晶体结构、相近的原子间距和热膨胀系数。利用界面合金、外延生长、真空淀积等技术,都可以制造异质结。各自合成后吸附的制备方法是可行的:双电极分别剥离两种材料,然后再在界面处进行自组装得到异质结,制备方法简单易行,并且利用电化学剥离的方法得到的异质结的尺寸较大,质量较高【5】. 另外,将一种在另一种生长上去叫是界面合成方法,两种同步合成的就是一锅粥的方法了。总之,三种方法都是可行,只有得到的产品满足“异质结”的特性要求即可。
问题2:请问制备的复合材料在反应的过程中分离了怎么办,有没有让两者结合紧密的方法,比如我想让硒化铜负载到钨酸铋上用什么方法比较好?
在传统材料学上,复合材料界面的作用,是在复合材料受到载荷时把基体上的应力传递到增强体上。这就需要界面相有足够的粘接强度,而两相表面能够互相浸润是先决条件。对于纳米结构的复合材料,强调的更多的是界面的化学键力和晶面匹配。纳米复合材料在应用过程中要求电子界面传导和物质扩散,三维多孔复合结构最为优先选择。复合材料在反应的过程中分离是很正常的事情,因为并不是所有的地方都形成真正的复合界面结构,有部分是物理的接触和堆积。大部分的文献可能表征的都是局部完美的复合结构。硒化铜负载到钨酸铋一般是水热加高温处理方法比较稳妥。
【1】科技名词——纳米复合材料 (baidu.com)
【2】第6章异质结2011 - 百度文库 (baidu.com)
【3】半导体异质结类型以及什么是能带对齐?
https://mp.weixin.qq.com/s/1zPrti1ez7n8q27xLBwWkg
【4】余家国最新Chem:新一代异质结——S型异质结光催化剂的那些故事
https://mp.weixin.qq.com/s/HZtSS8hA0kzlwCSKDcEiaQ
【5】一种异质结的制备方法 (xjishu.com)
【2】小知识 || Z-scheme异质结的类型与表征
Z-scheme异质结是指二者或二者以上的半导体接触以后,在光的激发下,对应半导体上导带(CB)和价带(VB)激发的电子和空穴分别迁移到相应另一半导体的VB和CB复合,使得二者半导体具有较强还原和氧化能力的电子和空穴得以保持,这样所构成的异质结我们称为Z-scheme异质结,这主要是由于其电子和空穴的传输途径类似于“Z”字母而命名的[1]。
Z型异质结的分类主要可分为以下三中,如图1-3[2]。其中图1表示的是液相间接Z-型异质结,主要是在液相中,含有能够转化电子和空穴的离子对,能够将半导体Ⅰ(SC Ⅰ)和半导体Ⅱ(SC Ⅱ)上的电子和空穴进行还原和氧化,达到传输电荷的作用,使得SC Ⅰ和SC Ⅱ上的空穴和电子得以保留,进一步与相应的物质发生氧化/还原反应。
图2表示的是全固态Z型异质结,在二者半导体中间具有高导电性的导电材料复合而成,在光激发以后,SC Ⅱ上的电子通过导电材料传输至SC Ⅰ,并与其对VB上的空穴发生复合,使得SC Ⅱ和SC Ⅰ上的空穴和电子得以保留。
图3表示的是直接Z型异质结,从图可以看出该异质结在光激发以后,SC Ⅱ上的电子可以直接与SC Ⅰ上的空穴发生复合,进而保留SC Ⅱ和SC Ⅰ上的空穴和电子。


图2 全固态Z-scheme异质结。

图3 直接Z型异质结
1. 由在反应过程中的活性物中的种类所对应的电势,通过EPR(电子顺磁光谱)以及光致发光捕获试验证明;
2. 通过KPFM(开尔文探针力显微镜)、表面光电压光谱学和瞬态吸收光谱学(TAS)测量半导体的表面电势;
3. 原位辐照X光电子能谱(ISIXPS)。在已建立的复合材料中,光生电子的迁移路径可以通过比较光照射前后组成元素的结合能位移来阐明。基于电荷迁移路线,可以评估直接Z方案电荷载流子转移;
4. 有效质量计算。根据密度泛函理论(DFT)计算,在相同条件下,有效质量较小的电荷载体具有较高的转移速率。因此,这预测了电荷载流子转移的趋势,以分析直接Z型异质结;
5. IEF(内建电场)评价。根据上述讨论,可以基于形成的IEF的方向来预测构建的光催化剂内的电荷转移路径。
参考文献:
[1]. Small Methods. 2017, 1,1700080
[2]. Angew. Chem. Int. Ed. 2020, 59, 22894–22915
https://mp.weixin.qq.com/s/A_prG8PQJQ5eYxGJhZUvHQ
【3】陕西科技大学谈国强课题组CEJ:全光谱广谱性降解抗生素的BiVO4@BiOCl晶面S异质结和Z型异质结

第一作者:杨迁
通讯作者:谈国强
DOI:10.1016/j.cej.2023.143450
图文摘要

成果简介
基于暴露晶面的BiVO4全光谱广谱性催化活性较差的问题,采用光照沉积晶面选择性诱导法,在BiVO4 (010)晶面异相成核形成强还原能力的BiOCl,在BiVO4 (110)晶面均相成核形成OVs-BiOCl,制备了核壳结构BiVO4@BiOCl晶面S和Z型异质结光催化剂。在表面异质结内建电场、界面静电内建电场、异质结间极化电荷转移和氧空位的LSPR效应的协同作用下,异质结光催化剂在全光谱范围具有良好的降解循环稳定性和较强的广谱性降解能力。在可见光/近红外光照射180 min后,异质结光催化剂对40mg/L TC的降解率分别为90.32%/71.17%,TOC去除率达到74.07%/57.81%,对CIP、BHA、苯酚、BPA和香豆素降解率分别为49.36~71.32%/37.88%~62.86%,且对混合抗生素溶液也具有良好的降解性能。本工作为表面异质结晶面选择性诱导氧空位促使异质结光催化剂增强的全光谱广谱性降解性能应用于环境净化领域提供了一条可行的研究思路。

图1. BiVO4@BiOCl异质结光催化剂的XRD图: (a)全谱图; (b)24°~28°放大图; (c)32°~34°放大图。
由图1(a)可知,单斜相BiVO4(BVO) (JCPDS编号14-0688) (110)、(121)、(010)、(200)和(002)晶面的衍射峰分别出现在2θ = 18.67、28.95、30.55、34.49和35.22处。四方相BiOCl(JCPDS 82-0485)(101)、(110)、(102)、(005)、(213)和(221)晶面的峰分别出现在2θ=25.91、32.55、33.52、63.14、65.81和69.53处。随着HCl浓度的升高,BiVO4@BiOCl样品中BiOCl的(101)、(110)、(102)晶面的峰相对强度逐渐增大(图1(b-c)),表明成功制备了核壳结构的BiVO4@BiOCl异质结光催化剂。

图2.BiVO4@BiOCl异质结光催化剂的微观形貌: (a)BVO; (b)BVO @ BiOCl-1; (c) BVO@BiOCl-2(插图:(110)晶面的局部放大图); (d)BVO @ BiOCl-3; (e)BVO @ BiOCl-2的EDS面扫图; (f)BVO@BiOCl-2的HRTEM图。
暴露(010)和(110)晶面的单斜相BVO具有光滑平整且相对锐利的边缘(图2(a))。较小的纳米片状的BiOCl紧密负载包裹十面体BVO的(010)晶面,团聚的不规则纳米颗粒状的BiOCl紧密负载在十面体BVO的(110)晶面上形成BVO@BiOCl-1 (图2(b))。在BVO@BiOCl-2异质结中,随着HCl溶液添加量的增加,牢牢结合在BVO(010)晶体面上的纳米片状BiOCl逐渐生长为片状BiOCl,纳米颗粒状的BiOCl紧密负载在十面体BVO的(110)晶面上(图2(c))。BVO@BiOCl-3异质结中,随着HCl溶液的不断增加,包裹在十面体BVO的(010)晶面的纳米片状BiOCl进一步生长,而负载在BVO的(110)晶体面上的纳米颗粒状BiOCl也逐渐生长成纳米片(图2(d)),形成了核壳结构的BiVO4@BiOCl异质结光催化剂。从核壳结构BVO@BiOCl-2的EDS图中可以发现,十面体BVO的(010)晶面Bi、V、O、Cl元素均匀分布,在BVO的(110)晶面中,Bi、V、O、Cl元素均匀地分布在上面两个晶面上,而在(110)晶面中,下面两个晶面上缺少O元素(图2(e)),表明BVO(110)晶面团聚的不规则纳米颗粒状BiOCl存在氧空位。在BVO@BiOCl-2异质结的HRTEM图中,0.255 nm、0.260 nm、0.292 nm和0.308 nm的晶格条纹间距分别对应BVO的(002)、(200)、(010)和(121)晶面。间距为0.135 nm、0.147 nm、0.267 nm、0.275 nm、0.142 nm和0.344 nm的晶格条分别对应BiOCl的(221)、(005)、(102)、(110)、(213)和(101)晶面(图2(f)),进一步表明BVO和BiOCl共存,其中团聚的不规则纳米颗粒状的BiOCl与BVO形成界面,进而形成紧密接触的异质结光催化剂。

图3. BiVO4@BiOCl异质结光催化剂EDS点扫描图像。
此外,在BiVO4@BiOCl (010)晶面上取4点进行EDS检测,发现各点均存在Bi、V、O、Cl元素。对BiVO4 @ BiOCl)晶面上八个点的EDS测试表明,每个点上也存在Bi、V、O和Cl。(110)晶面上的八个点都比(010)晶面上的四个点含有更少的氧元素(图3),这进一步表明了氧含量的不同表面。

图4. BiVO4@BiOCl异质结光催化剂的形成机理。

图5. 功函数:(a)BiVO4 (110)/BiVO4(010); (b)BiOCl; (c)BiVO4@BiOCl。
结合DFT模拟,进一步验证了BiVO4@BiOCl异质结的形成机理,如图4所示。由于BVO的(010)和(110)晶面形成表面异质结(图4(a)),在BVO溶液的UV光激发之后,表面异质结能级差异导致h+和e-分别输运和聚集到BVO的(110)和(010)晶面。e-从BVO (010)晶面迁移到(110)晶面,抬升BVO (110)晶面的Ef BVO (110),直到两个晶面的费米能级平衡(图4(b)中Ef BVO (110)/ BVO (010)的蓝色虚线)。同时,在从(010)晶面指向(110)晶面的BVO (010)晶面和BVO (110)晶面之间产生了内置的界面电场E1(图4(b))。Bi(NO3)3∙5H2O溶于乙二醇溶液中,由Bi3+和乙二醇生成带正电的醇氧化合物[BiOCH2CH2OH]2+。当加入到UV照射后的BVO水溶液中时,带正电的[BiOCH2CH2OH]2+通过静电引力被吸附到BVO (010)晶面上。滴加HCl溶液后,[BiOCH2CH2OH]2+通过与HCl在BVO(010)晶面上发生异相成核反应形成片状BiOCl (图4(b))。同时,在BVO (010)晶面和BiOCl之间产生静电内建界面电场E2,方向从BiOCl指向BVO (010)晶面。计算出BVO (110)/ BVO (010)的功函数(Wf)为4.73 eV(图5(a)),而BiOCl的Wf为7.38 eV(图5(b))。由于遵循异相成核,在功函数的作用下,电子倾向于从BVO的Ef BVO移动到BiOCl的Ef BiOCl,进一步抬升BiOCl的Ef BiOCl,直到两个费米能级平衡(图4(c)中Ef BVO-BiOCl的红色虚线)。BVO-BiOCl的Wf为6.29 eV(图5(c)),导致BiOCl的费米能级和能带位置提升约1.09 eV,CB/VB势分别达到-1.358 eV/1.723 eV(图4(c))。此外,在滴加HCl溶液的过程中,Cl-通过静电引力吸附在带正电的BVO (110)晶面上,利用BVO (110)晶面上的Bi3+作为Bi源,在HCl和H2O的存在下,通过在BVO (110)晶面上的原位水合反应均相成核,形成含OVs的BiOCl。同时,BVO (110)晶面与OVs-BiOCl之间形成内建界面电场E3,方向从BVO (110)晶面指向OVs-BiOCl(图4(d))。由于发生均匀形核反应,因此OVs-BiOCl和(110)晶面上的BVO费米能级不会随功函数而改变。在表面异质结内置电场、内置界面电场静电形成以及功函数差异导致的极化电荷转移的作用下,在BVO (010)晶面上异相成核形成了BVO和BiOCl的晶面S型异质结,极化电荷转移提高了BiOCl的能带结构,增强了BiOCl的还原能力。在BVO (110)晶面上由于均相成核形成OVs-BiOCl,并在静电内建电场作用下,形成BVO (110)晶面Z型异质结。因此,利用均相成核原理选择性诱导OVs-BiOCl在BVO (110)晶面上的形成,从而通过光照沉积晶面选择性诱导法制备了含有晶面OVs的核壳结构BiVO4@BiOCl晶面S型和Z型异质结光催化剂。
利用四环素(TC)作为基准,评估每个样品的光催化效率。光照前先在黑暗环境下进行暗吸附,使反应体系达到吸附-解吸平衡。各样品在可见光照射下对TC的降解性能如图6所示,TC的自降解效率极低且不显著。在可见光下反应180 min后,BVO、BVO@BiOCl-1、BVO@BiOCl-2和BVO@BiOCl-3对40 mg/L TC的降解率分别为50.16%、79.16%、90.32%和73.49%(图6(a)),其反应的表观速率常数分别为0.00394 min-1、0.00811 min-1、0.0118 min-1和0.00742 min-1(图6(b))。BVO@BiOCl-2对40 mg/L TC的TOC去除率为74.07%,表明其具有优异的矿化能力(图6(c))。在可见光照射下循环四次的降解率分别为90.32%、90.14%、89.39%和88.27%,降解效率仅仅下降2.05%(图6(d)),表明其在可见光条件下具有良好的循环稳定性。

图6. BiVO4@BiOCl异质结光催化剂在可见光照射下对TC的降解反应:(a)TC降解曲线; (b)动力学拟合曲线; (c)TOC去除率; (d)循环试验。
通过在近红外光下降解TC评估了BiVO4@BiOCl异质结的全光谱催化效率。BVO、BVO@BiOCl-1、BVO@BiOCl-2和BVO@BiOCl-3对40 mg/L TC的降解率分别为0%、64.41%、71.17%和62.66%(图7(a))。在近红外光下,BVO对TC无降解作用,而BiVO4@BiOCl异质结光催化剂的光催化效率显著提高。各样品的TC表观降解速率常数由小到大依次为BVO@BiOCl-3 (0.00509 min-1)<BVO@BiOCl-1 (0.00544 min-1)<BVO@BiOCl-2 (0.00661 min-1)(图7(b))。BVO@BiOCl-2在近红外光照180 min后,对40 mg/L TC的TOC去除率达到57.81%(图7(c)),表明其在近红外光下也具有良好的矿化能力。在近红外光下四个循环的降解率分别为74.16%、73.86%、72.39%和70.46%(图7(d)),降解率仅下降3.70%,这表明在近红外光条件下同时具有良好的循环稳定性。

图7. BiVO4@BiOCl异质结光催化剂在近红外光照射下对TC的降解反应:(a)TC降解曲线; (b)动力学拟合曲线; (c)TOC去除率; (d)循环试验。
为了探索BiVO4@BiOCl异质结光催化剂在全光谱范围内的广谱性能,我们进行了在可见光/近红外光下对CIP、BHA、苯酚、BPA和香豆素的降解实验。10 mg/L的CIP、BHA、苯酚、BPA和香豆素在可见光下180 min的降解效率分别为71.32%、49.36%、65.69%、57.20%和57.52%。在近红外光下照射180 min的降解效率分别为37.88%、48.22%、62.86%、53.48%和55.45%(图8(a)),表明制备的BiVO4@BiOCl异质结在整个光谱范围内都具有很大的广谱降解能力。其对CIP、BHA、苯酚、BPA和香豆素的TOC去除率分别达到54.58%/49.15%、32.78%/28.51%、53.03%/48.96%、37.24%/32.64%和35.65%/32.99%(图8 (b))。结果表明,BiVO4@BiOCl异质结光催化剂在全光谱范围内具有优良的广谱矿化能力。
将TC (40 mgL-1)、CIP (20 mgL-1)和BHA (10 mgL-1)三种抗生素混合,进一步评价样品的实际适用性。分别得到了BiVO4@BiOCl异质结光催化剂在可见光和近红外下降解混合抗生素溶液的紫外-可见光谱。随着降解时间的延长,吸收带逐渐变弱,光反应180 min后吸收带基本消失(图8(c-d)),证明BiVO4@BiOCl异质结光催化剂对混合抗生素溶液具有良好的降解性能。

图8. BiVO4@BiOCl异质结光催化剂对CIP、BHA、苯酚、BPA和香豆素的降解反应: (a)可见光和近红外光下的降解率; (b)可见光和近红外光下的TOC去除率; 混合抗生素降解的紫外-可见光谱图: (c)可见光; (d)近红外光。
图9(a)显示BiVO4@BiOCl异质结光催化剂的紫外可见漫反射光谱。BVO的特征吸收边大约在500 nm处,在BVO不同晶面负载BiOCl进行改性后的BVO@BiOCl异质结在200~2200 nm的全光谱范围表现出的光吸收明显比BVO增强,这是由于BiOCl的存在提升了BVO可见光的利用,同时BVO (110)晶面选择性诱导形成OVs-BiOCl,OVs的LSPR效应拓宽了异质结在780~2200 nm近红外光区域范围的光吸收能力。根据DRS结果,可以用公式(αhν)2=A(hν-Eg)n确定BVO带隙值。对于n=1的直接跃迁型半导体BVO, BVO的能带值为2.356 eV(图9(a)插图)。图9(b)为黑暗条件下进行的Mott-Schottky实验,可以看出BVO具有n型半导体的特性。通过线性与x轴的截距,计算出BVO的CB电势约为-0.351 eV, BVO的VB电势约为2.005 eV,这与BVO的XPS-VB光谱一致(图10(a))。BiOCl带隙值可由公式(αhν)1/2=A(hν-Eg)n确定,得到的BiOCl能带值为3.081 eV(图10(b))。同时,从BiOCl的XPS-VB光谱中得到VB电位约为2.813 eV(图10(c)),结合ECB=Eg-EVB得到BiOCl的CB电位约为-0.268 eV。

图9. BiVO4@BiOCl异质结:(a)紫外-可见漫反射图(插图:光子能量图); (b)Mott-Schottky曲线; 活性物种捕获实验: (c)可见光下; (d)近红外光下。
加入BQ、Na2C2O4和TBA后,其在可见光下的光催化性能分别下降了51.64%、16.02%和28.82%。这表明在可见光降解过程中,O2‑是主要的活性物种,其次是OH和h+(图9(c))。在近红外光照射条件下,加入BQ、Na2C2O4和TBA后,光催化剂的降解能力分别从71.17%下降到26.58%、66.33%和38.35%(图9(d))。近红外光降解过程中,O2‑是降解过程中的主要活性物质,而OH和h+在降解过程中起次要作用。

图10. XPS-VB光谱: (a)BVO; (b)BiOCl; (c) BiOCl的DRS光谱。
基于以上结果可以推测出BiVO4@BiOCl异质结光催化剂的全光谱光催化增强机理(图11)。BiVO4@BiOCl异质结光催化剂在可见光光照下,BVO和BiOCl光响应产生光生电子和空穴。在BVO (010)晶面和BVO (110)晶面界面处内建电场E1作用下,BVO (110)晶面光生电子迁移到BVO (010)晶面上,BVO (110)晶面迁移到BVO (010)晶面上的光生电子和BVO (010)晶面光激发产生的光生电子在BVO (010)晶面与BiOCl界面形成的静电内建电场E2的作用下,与BiOCl价带上光生空穴在界面处复合,在(010)晶面BVO与BiOCl在BVO的(010)晶面处形成了晶面S型异质结,保留了BiOCl导带上光生电子。BiOCl的导带电势为-1.358 eV,因此BiOCl导带上所保留的光生电子与溶液中溶解O2形成为•O2‑自由基,参与降解反应(图11(a))。此外,BVO (010)晶面价带上光生空穴在内建电场E1作用下迁移到BVO (110)晶面价带上,在BVO (110)晶面与OVs-BiOCl的界面形成的内建电场E3的作用下,OVs-BiOCl导带上的光生电子与BVO (110)晶面价带上光生空穴和BVO (010)晶面价带上迁移到BVO (110)晶面价带上光生空穴一起在界面处复合,保留了OVs-BiOCl价带上的光生空穴,OVs-BiOCl与(110)晶面BVO在BVO的(110)晶面形成了晶面Z型异质结,OVs-BiOCl价带上所保留的空穴电势(+2.813 eV)高于•OH自由基的生成电势(EH2O/•OH=+2.40 eV,EOH-/•OH=+1.99 eV),产生的•OH自由基参与降解有机污染物,或保留的h+直接降解有机污染物,这与可见光的活性物种测试相符。
在近红外光照射下,由于较低的光子能量,BVO和BiOCl在近红外光照射下均无法被激发。但BVO (110)晶面上选择性诱导形成的OVs-BiOCl中的氧空位在近红外光照射下具有明显的LSPR效应,其费米能级周围的自由载流子可与入射光子相互作用,产生高能热电子;高能热电子与溶液中溶解O2形成为•O2‑自由基(图11(b)),直接降解有机污染物。同时氧空位的LSPR效应产生局域磁场的共振促使BVO和BiOCl吸收近红外光(图11(b)中绿色波浪线),致使BVO和BiOCl产生光响应产生光生电子和空穴,然后遵循BVO与BiOCl在(010)晶面和(110)晶面界面形成的晶面S和Z型异质结机制进一步降解有机污染物,这与近红外光下的活性物种测试相符。

图11. BiVO4@BiOCl异质结光催化剂全光谱光催化增强机理图: (a)可见光; (b)近红外光。
利用ESR自旋捕获光谱进一步佐证BiVO4@BiOCl异质结光催化机理。在黑暗条件下均无法观察到•O2−、•OH和1O2特征峰信号。可见光照射10 min和20 min后均显示出DMPO-•O2−加合物、DMPO-•OH和TEMP-1O2的特征峰,并且强度随着时间的延长而增加(图12(a-c))。近红外光照射10 min和20 min后结果中也发现了代表•O2−、•OH和1O2的特征峰(图12(d-f))。由此可知,在BiVO4@BiOCl异质结光降解TC的过程中,有活性物种•O2−、•OH和1O2参与了降解的氧化还原反应。1O2特征峰也进一步证明了在BVO的(010)晶面和(110)晶面发生了电子和空穴的复合形成了激子(e--h+)(图10(b)中黄绿色箭头),进一步验证了核壳结构BiVO4@BiOCl晶面S和Z型异质结机制的合理性。

图12. BiVO4@BiOCl异质结光催化剂的EPR图谱,可见光: (a)DMPO-•O2−; (b)DMPO-•OH; (c)TEMP-1O2; 近红外光: (d) DMPO-•O2−; (e)DMPO-•OH; (f)TEMP-1O2。
采用光照沉积法,以暴露(010)和(110)晶面的十面体单斜相BiVO4为内核,在BiVO4的(010)晶面上通过异相成核形成片状四方相BiOCl,在BiVO4的(110)晶面上通过均相成核形成颗粒状四方相OVs-BiOCl,制备了核壳结构BiVO4@BiOCl晶面S和Z型异质结光催化剂。在可见光/近红外光照射180 min后,异质结光催化剂对40mg/L TC的降解率分别为90.32%/71.17%, TOC去除率达到74.07%/57.81%,对TC的降解作用是真实的矿化降解而不是物理吸附。在可见光和近红外光照射180 min后,对CIP、BHA、苯酚、BPA和香豆素降解率分别为49.36~71.32%和37.88%~62.86%,且对TC、CIP和BHA混合抗生素溶液也具有良好的降解性能。在表面异质结内建电场、界面静电内建电场、功函数差异引起的异质结极化电荷转移和氧空位LSPR效应的协同作用下,增强了异质结光催化剂在全光谱范围良好的降解循环稳定性和较高的广谱降解能力。本工作为表面异质结晶面选择性诱导氧空位制备全光谱响应广谱性降解抗生素的异质结光催化剂应用于环境净化领域提供了一条可行的研究思路。
https://mp.weixin.qq.com/s/DgLxW4EStMNBuIilV39hzg
【4】余家国Angew:利用飞秒瞬态吸收光谱验证S型异质结中的电荷转移机制


【研究背景】


【成果简介】


【研究亮点】


【图文导读】







【总结与展望】


【文献链接】

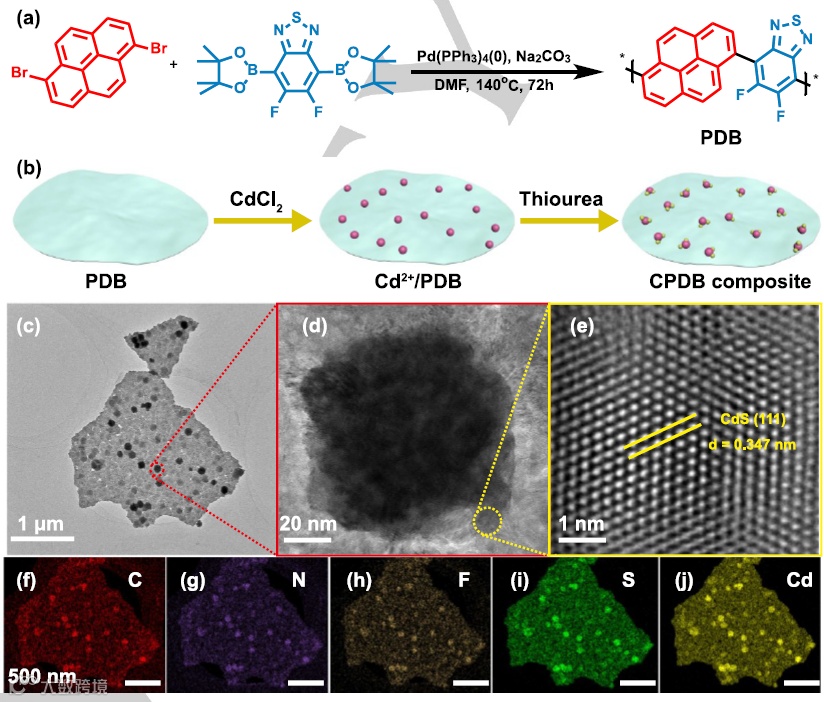
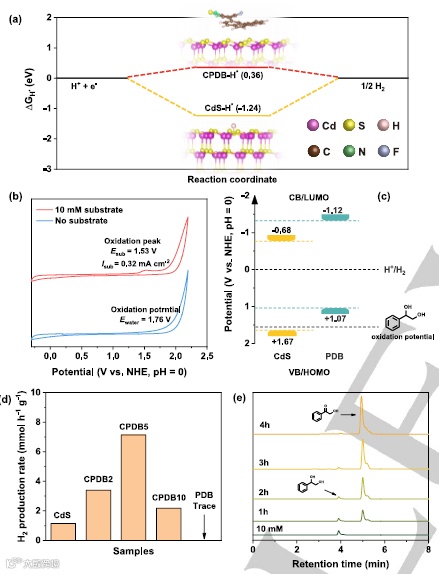
【5】双S型异质结光催化产氢新进展!

吉林大学/江苏大学/香港城市大学|崔小强/姜志峰/Sai Kishore Ravi|Adv. Mater.|一种具有自组装异质结的双S型人工光合系统产生优异的光催化析氢速率

1
文章信息

一种具有自组装异质结的双S型人工光合系统产生优异的光催化析氢速率
通讯作者:崔小强/姜志峰/Sai Kishore Ravi
通讯单位:吉林大学/江苏大学/香港城市大学等
Doi:10.1002/adma.202209141
2
研究背景

在传统的Z方案异质结中,光催化剂的界面接触较差,严重阻碍了有效的电荷输运。由还原型和氧化型光催化剂组成的S型(即步骤型)异质结被认为是一种很有前途的提高光催化性能的策略。与传统的S型光系统相比,Z型结构引起的内部电场、带弯曲和库仑吸引可以促进电荷转移,提高光催化性能。虽然S型结构在增强光催化性能方面具有潜在的优势,但单结S方案结构仍存在一些缺陷,如相互作用弱、多相集成弱等。人们已经尝试通过在体系中使用三种不同的催化剂来创建一个额外的结来克服这一点。然而,三种不同催化剂的界面接触差和长期载流子转移机制模糊仍然是一个瓶颈。为了解决这个问题,我们提出了一种新的“双S方案”结构。具有双S型机制的光催化剂包括两种氧化型光催化剂(OP)和一种还原型光催化剂(RP)。与之前发表的关于双S型结构的工作相比,值得注意的是,我们在双S型结构中选择的氧化型光催化剂是相同的半导体不同的相。我们假设,使用不同氧化型异相光催化剂可以更好地与还原型光催化剂能量对齐,增强界面相互作用。在没有光照射的情况下,当RP和OP接触时,结处出现了能带弯曲和内电场(IET)。然而,当暴露于光照下时,RP和OP分别形成富电子区和富空穴区。在内部电场(IET)下,OP的导带(CB)上的光生电子被驱动来消耗RP的价带(VB)上的空穴。然后将有意义的光电子和空穴分别积累在RP的CB和OP的VB上,使异质结具有最高的氧化还原能力。与单结S型结构相比,双S型结构可以在光催化剂之间产生两个内部电场,从而产生更有效的空间电荷转移和更强的氧化还原能力。虽然双S方案结构有两个连接,但我们只选择了一种具有不同异相的OP与一种RP连接。该设计克服了传统三元异质结具有高接触势垒、晶格匹配差和界面相互作用的局限性。
3
文章简介

基于上述现状,吉林大学/江苏大学/香港城市大学等|崔小强/姜志峰/Sai Kishore Ravi课题组在《Adv. Mater.》上发表了题为A Twin S-Scheme Artificial Photosynthetic System with Self-Assembled Heterojunctions Yields Superior Photocatalytic Hydrogen Evolution Rate的研究文章。该课题组报道了一种自组装结构,将二维石墨氮化碳纳米片插入锐钛矿二氧化钛纳米颗粒和H掺杂金红石二氧化钛纳米棒之间,产生了双S型光催化剂(表示为TSP)。该TSP催化剂在紫外-可见光照射下表现出前所未有的光催化析氢速率62.37 mmol g−1h−1,表观量子效率为45.9%。通过x射线光电子能谱(XPS)、电子自旋共振(ESR)自旋捕获试验和带结构分析,确定了双S型电荷转移路径。飞秒瞬态吸收(fs-TA)光谱、瞬态表面光电压(TPV)和非原位表征进一步证明了新型双S方案机制对光生载流子的有效空间分离和转移。这项工作为设计具有S型电荷转移路径的光催化性能的异质结提供了一个新的视角。
4
图文解析


图1.a)自然光合作用中的Z方案,b)人工光合系统中的Z方案,c)S方案,d)双S方案的人工光合系统在接触前后,以及在光照下。e)材料的静电自组装示意图。
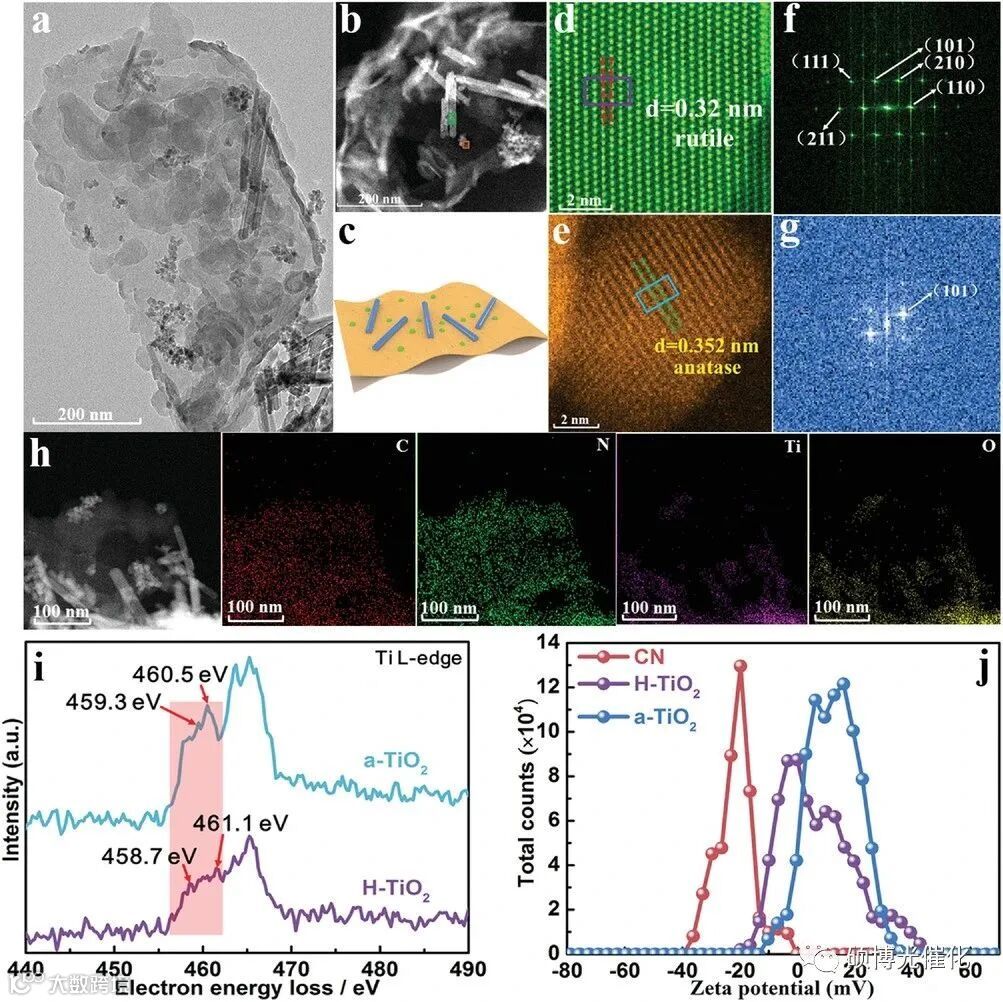
图2.TSP异质结的a) TEM图像。b)TSP异质结的HAADF-STEM图像。c)由C3N4纳米片(橙色)、金红石二氧化钛纳米棒(蓝色)和锐钛矿二氧化钛纳米颗粒(绿色)组成的TSP异质结示意图。d,e)绿色矩形和棕色正方形的放大图像。f,g)分别从相应的绿色矩形(金红石纳米棒)和棕色正方形(锐钛矿纳米颗粒)中拍摄的FFT图像。h)TSP的STEM元素映射。i)绿色区域的H-TiO2和棕色区域的锐钛矿TiO2的EELS光谱。j)原始CN、H-TiO2和a-TiO2的Zeta电位。

图3.a)TSP与商业金红石、C3N4、锐钛矿、P25、H-TiO2、aTiCN、HTiCN和NT-TiCN在紫外-可见照射(波长范围为200-1000nm)下的光催化H2演化速率。b)TSP的外部表观量子效率。c)TSP在紫外-可见光下的循环实验(注:催化剂溶液密封,在黑暗中保存半个月)。d)与之前报道的TiO2基催化剂相比,TSP实现了光散分解太阳能H2演化速率(HER)。误差条表示三次重复的平均±标准差.为e)紫外和可见波长的氙气灯照射TSP的照片。

图4.a-TiO2、H-TiO2、CN、TSP的a) Ti 2p、c、N 1s的x射线光电子能谱(原位和原位)光谱。DMPO-超氧自由基的CN、H-TiO2、a-TiO2、HTiCN、aTiCN和TSP e)和DMPO-羟基自由基在水分散5 min时的电子自旋共振波谱(ESR)信号。TSP g)和5、10、15、30 min光照射下的原位ESR光谱
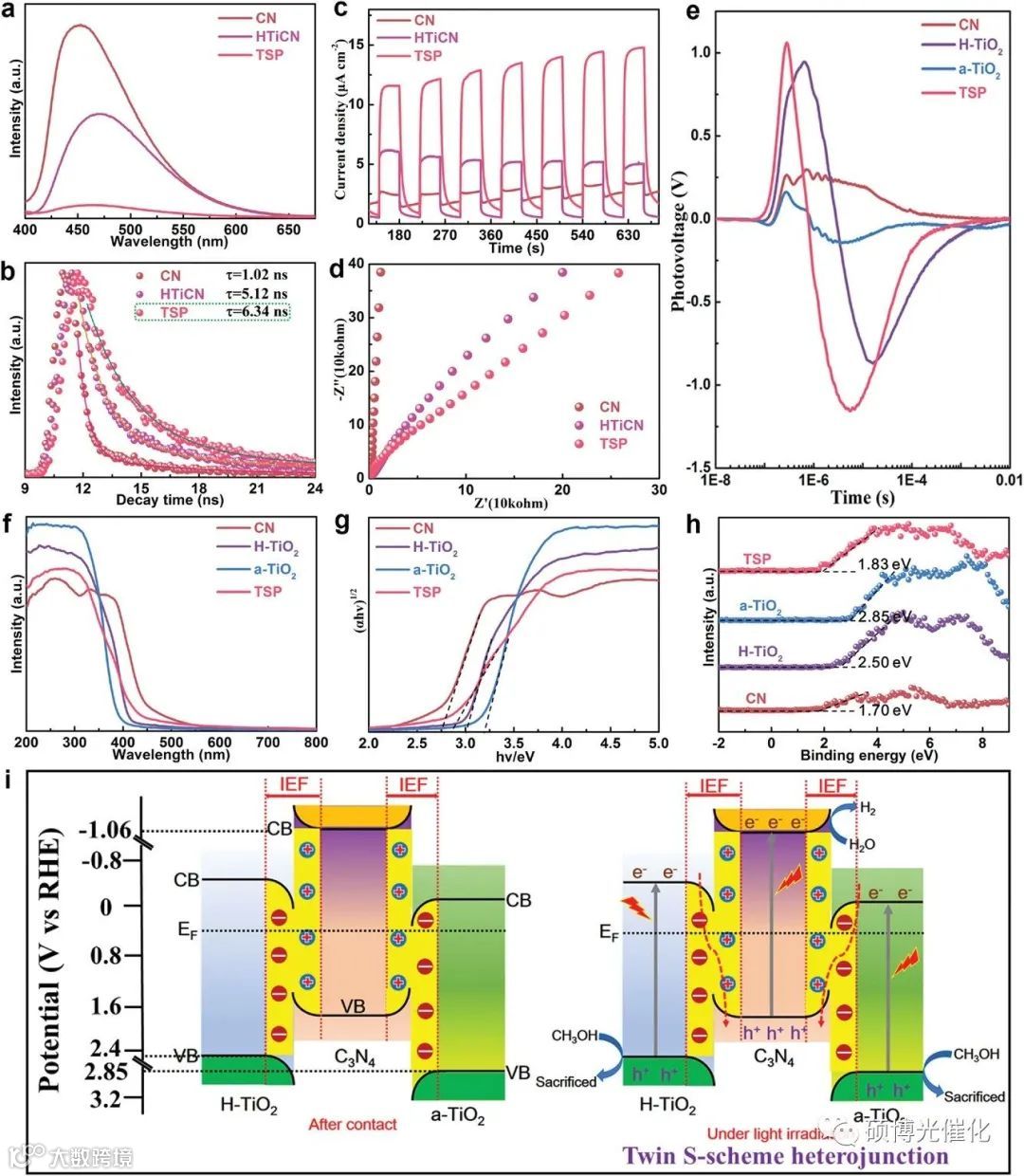
图5.a)光致发光光谱。b)时间分辨荧光光谱,c)瞬态光电流响应和d)CN、HTiCN和TSP的电化学阻抗光谱(EIS)。CN、H-TiO2、a-TiO2和TSP的瞬态表面光电压(TPV)。f)CN、H-TiO2、a-TiO2和TSP的紫外-vis DRS。g)使用(F(R)hv)1/2=作为光子能量的函数,为CN、H-TiO2、a-TiO2和TSP绘制的相应Tauc图。h)CN、H-TiO2、a-TiO2和TSP的VB-XPS光谱。CN、a-TiO2和H-TiO2的VB最大值符合光电效应方程。i)双S型异质结的能带对准。

图6. a、b)CN、d、e)H-TiO2、g、h)a-TiO2和j、k)TSP催化剂的瞬态吸收光谱。c) CN、f) H-TiO2、i) a-TiO2和l) TSP催化剂在400 nm激光脉冲照射后的归一化瞬态吸收时间曲线。
https://mp.weixin.qq.com/s/JlrIlm2S3z22xP_zZYEeKw