
摘要:
研究结果
1.研究设计
我们的研究设计结合了广泛的联盟数据、内部正常组织混合物RNA-seq数据以及一名代表癌症患者的ctcRNA-seq数据。全面的研究设计和概念框架如图 1 所示。为确保研究结果的稳健性,我们进行了三个不同的验证程序。第一项验证涉及半硅学方法,第二项验证利用了由六种组织类型组成的组织混合物的独立RNA-seq分析。最后,利用一名转移性肿瘤患者的纵向ctcRNA-seq数据进行了最终的独立验证。这一最终验证体现了早期检测转移性肿瘤的潜在临床应用,并突出了我们研究的转化意义。在验证过程中,我们对深度学习方法和NNLS方法进行了全面比较,凸显了深度学习方法在各种情况下表现出的一致优势。

The overall study designs. The neural network deep learning model was trained using GTEx RNA-seq data from TSGs identified in GTEx and TCGA normal tissue. The performance of the model was compared to the traditional NNLS method in three independent validation datasets: (1) Semi in silico dataset from GTEx; (2) Semi in silico dataset from six normal tissue RNA-seq; (3) ctcRNA-seq from a melanoma patient with metastatic tumors
2.组织特异性基因(TSG)
我们选择了 15 种组织类型(脑、乳腺、结肠、食道、肾、肝、肺、卵巢、胰腺、前列腺、皮肤、小肠、胃、甲状腺和子宫)。之所以选择这些组织类型,是因为它们有现成的 RNA-seq 数据,而且是常见的癌症部位。为了确定组织特异性基因(TSGs),我们首先利用了GTEx RNA-seq数据集。随后,我们用TCGA的正常组织 RNA-seq 数据集验证了这些 TSG。例如,在GTEx RNA-seq数据中发现了仅存在于小肠的TSG MAP4K1,并通过 TCGA 正常组织 RNA-seq 数据的验证进一步证实了这一点(图 2A,B)。验证过程可确保观察到的组织特异性不受来自单一数据集的噪音或批次效应的影响。最终,我们的组织特异性标准共鉴定出6,558个符合既定标准的基因。在所考虑的组织类型中,每个组织的 TSG 平均数量为642个。大脑中的组织特异性基因(TSG)数量最多,共有1853个,而胃中的 TSG 数量最少,只有163个(图 2C)。

Illustration of our TSG analyses. A Boxplots show that the gene MAP4K1 is primarily expressed only in the small intestine in GTEx. B Boxplots show that the gene MAP4K1 is primarily expressed only in the small intestine in normal tissues in TCGA which confirms the finding in GTEx. C A Bar chart that depicts the number of TSGs in each tissue type. Large variations in the number of TSGs can be observed. D Pie charts that the results from gene stability tests. Gray color indicates the proportion of TSGs that showed no notable change between tumor and normal. Red color indicates the proportion of TSGs that showed upregulation in tumors. Green indicates the proportion of TSGs that showed downregulation in tumors. E–G shows results from independent validation of the single tissue deep learning prediction model using TCGA normal tissue RNA-seq data. E Confusion matrix that shows overall high accuracy of the single tissue deep learning prediction model. F A polygon plot that demonstrates performance of the single tissue deep learning prediction model in F1 score, precision, and recall. G ROC curves show the high performance of the single tissue deep-learning prediction model
接下来,我们在肿瘤样本中进行了TSG稳定性测试,旨在证明相当一部分已确定的组织特异性基因(TSG)不会受到肿瘤发生的影响。为此,我们利用TCGA RNA-seq数据进行了基因表达分析,将肿瘤样本与相应的正常组织样本进行了比较(图 2D)。不过,由于某些癌症类型无法获得邻近的正常组织,因此该分析仅限于 12 种组织类型。平均而言,约 50%的组织特异性基因(TSGs)在肿瘤样本和正常样本之间没有明显的表达差异,范围从肺癌的26%到胰腺癌的96%。相反,约25%的TSG在肿瘤样本中表现出上调,从胰腺癌的3%到肺癌的44%。此外,约有25%的TSGs在肿瘤中出现下调,从胰腺癌的1%到肺癌的35%不等。从正常状态向肿瘤状态过渡期间基因表达的失调可能会影响组织解卷积分析中TSG的有效性。不过,我们随后的分析表明,深度学习模型对基因表达的变化表现出更强的适应能力,减轻了这些失调的影响。
为了确定深度学习方法的可行性,我们首先利用GTEx数据集开发了一个专门用于单个组织预测的深度学习模型。正如预期的那样,在使用TCGA正常组织RNA-seq数据作为独立测试数据集进行评估时,该模型表现出了卓越的性能。总体准确率达到96%(图 2E)。此外,该模型的F1分数、召回率和精确度指标也表现出显著的性能水平(图 2F)。接收者操作特征曲线(ROC)进一步证实了这些发现,因为大多数组织类型的曲线下面积(AUC)值都接近 1,这表明该模型具有很高的判别能力(图 2G)。
3.通过半硅学数据进行验证
为了评估和比较我们的深度学习组织解卷积模型的性能,我们进行了三次不同的验证测试和比较分析。最初的验证方法是利用基因型-组织表达(GTEx)项目获得的RNA-seq数据生成的半硅学数据集。由于GTEx数据集包含来自个体捐献者的多个组织样本,因此特别适合用于半硅学 RNA 混合物。我们利用“方法”部分概述的方法构建了一个半硅学数据集,其中包括1000个随机组织混合物RNA-seq样本。这使我们能够模拟各种 RNA 混合物,并创建一个全面的数据集,用于对我们的深度学习模型进行严格的评估和验证。我们使用深度学习模型和NNLS方法对相同的半硅学数据集进行了解卷积。这些方法的功效通过两个性能指标来评估:皮尔逊相关系数和均方误差(MSE)。计算随机生成的组织贡献与1000个半硅学组织混合物样本中预测的组织贡献之间的相关性(r)。平均而言,深度学习模型的相关性为0.98,且变化较小(图 3A),而NNLS的平均相关性为0.79(范围:结肠为0.47,卵巢为0.97)(图 3B)。MSE是根据每个半硅学样本的随机组织贡献到预测回归模型的平均距离计算得出的。此外,还进行了敏感性分析,以评估缺失数据对两种方法的影响。在评估深度学习模型和NNLS方法时,随机选择了2000到6000个组织特异性基因(TSGs),间隔为500个增量。值得注意的是,深度学习模型在相关性(图 3C)和MSE(图 3D)方面始终优于NNLS方法。这一观察结果凸显了深度学习模型在应对实际应用中普遍存在的数据缺失所带来的挑战时的应变能力。
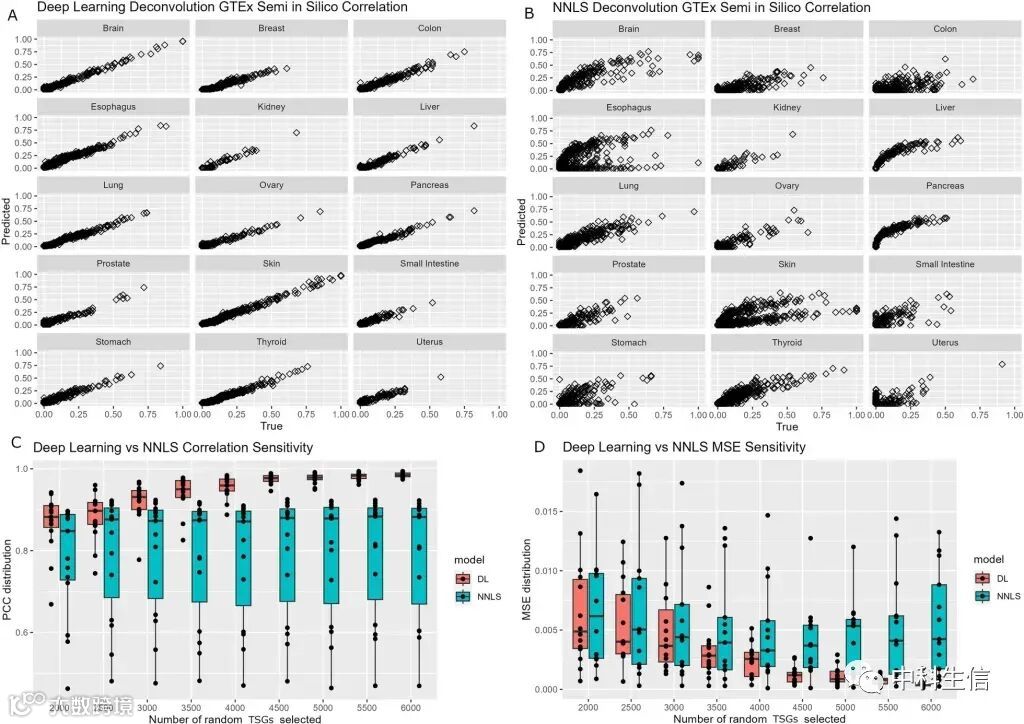
Data analyses illustrating validation using semi in silico GTEx data and comparison between deep learning model and NNLS method. A Scatter plots that depict the correlation between randomized tissue contribution and predicted tissue contribution by deep learning model. B Scatter plots that depict the correlation (r) between randomized tissue contribution and predicted tissue contribution by the NNLS method. C Boxplots that show higher performance in the sensitivity analysis by deep-learning model. D Boxplots that show smaller MSE in the sensitivity analysis by deep-learning model
4.正常组织混合物 RNA-seq 验证
之前利用半硅学GTEx数据进行的验证表明,深度学习模型的结果很有希望。不过,值得注意的是,尽管采用了随机化,GTEx数据集仍被用于训练和验证,因此缺乏完全的独立性,可能会引入偏差。为了解决这个问题,我们在六个不同的正常组织(脑、乳腺、结肠、肾、肝和肺)上进行了RNA-seq实验,这些组织来自新墨西哥大学的人类组织库。这种方法确保了与GTEx数据集的绝对独立性,增强了我们验证程序的可靠性和无偏性。
采用上述策略,我们利用从六个正常组织中获得的RNA-seq数据构建了一个包含1000个样本的半硅学数据集。该RNA-seq数据包含了之前发现的6558个 TSG中的4973个,切实地证明了实际应用中可能遇到的差异。
通过使用皮尔逊相关性和MSE作为性能指标,利用深度学习和NNLS方法评估了组织解卷积的性能。我们的深度学习模型表现出色,平均相关性达到0.97(在脑组织中为0.95,在肺组织中为0.98)。相比之下,NNLS方法的平均相关性为0.89(在乳腺组织中为0.71,在肾脏组织中为0.95)。我们进行了敏感性分析,以仔细检查这两种方法在不同情况下的性能,随机基因集从2000个TSG到6000个TSG,递增间隔为500。结果显示,深度学习模型的相关性(图 4A)和MSE(图 4B)均优于NNLS。对 1000 个半硅学样本的评估表明,深度学习模型的 MSE 值始终低于NNLS方法(图 4C)。总体相关性结果也有利于深度学习(图 4D)。这些令人信服的发现进一步证实了深度学习在组织解卷积领域的优势能力。

The results from validation by normal tissue RNA-seq data and patient ctcRNA-seq data. A A dot plot that shows higher correlation (r) achieved by deep learning model in the sensitivity analysis. B A dot plot that shows small MSE achieved by deep learning model in the sensitivity analysis. For A and B, the dotted line indicates the mean value. C A box violin and dot combination plot that shows smaller MSE for all 1000 semi in silico samples. D A dot plot that shows overall higher correlation by deep learning model. E A dot plot that shows the proportion of tissue contribution in 9 month follow up for a melanoma patient with brain and lung metastatic tumors. F A heatmap that depicts the complete 15 tissue results by deep learning model. G A heatmap that depicts the complete 15 tissue results by the NNLS method
5.利用 ctcRNA 进行组织解卷积
我们分析了一个纵向ctcRNA-seq数据集,该数据集取自一名肺部和脑部出现转移性肿瘤的黑色素瘤患者。ctcRNA是在三个不同的时间点采集的:ctcRNA是在最初诊断后的0、3和9个月收集的。利用深度学习和NNLS方法,我们努力辨别ctcRNA的组织来源(图 4E)。经过分析,我们发现这两种方法都能在0个月的时间点成功检测到皮肤组织的存在。然而,与NNLS方法相比,深度学习方法的皮肤检测比例更高。值得注意的是,在3个月和9个月的时间点,NNLS 方法未能识别出任何皮肤组织。此外,随着时间的推移,深度学习模型发现肺部和脑部组织的比例越来越高,这表明转移的进展。相比之下,NNLS方法只能发现肺组织比例的增加,却无法检测到脑组织比例的任何变化。图 4F是深度学习的结果,图 4G是NNLS的结果。值得注意的是,这两种方法都一致检测出乳腺、卵巢和子宫等女性器官的内容微乎其微或几乎没有,这与患者的男性性别相符。值得注意的是,与半硅学数据集不同,病人样本的精确地面实况仍然未知。因此,由于数据集的复杂性和固有局限性,在比较深度学习和NNLS方法时得出明确结论具有挑战性。尽管如此,观察到的结果仍然与根据患者的转移病史和性别推断出的总体趋势一致。研究结果与已知临床信息之间的这种一致性进一步凸显了所采用技术的潜在临床实用性。
总结
我们的研究结果凸显了在组织解卷积中使用深度学习模型的好处,包括其在验证数据集中的性能提升以及对缺失数据问题的适应能力。此外,我们还展示了ctcRNA在组织解卷积中的新应用,并提供了支持其可行性可靠性的证据。将深度学习模型与ctcRNA结合起来,对转移的早期检测、治疗决策、疾病监测、耐药性预测和生物标记物的发现等具有广泛的临床意义。利用深度学习对ctcRNA进行分析,有望促进我们对癌症生物学的理解,并加强对患者的护理。
