
蛋白质组差异分析:常用R包选择与实操指南
上一期我们掌握了数据归一化处理和PCA分析的代码。了解数据的整体分布之后,如何筛选差异蛋白?如何选择合适的差异分析R包?本期将从最常用的limma包入手,结合DEP2和DEqMS,构建适合蛋白质组学新手的实用分析指南。
本期聚焦:
- 转录组的R包可以用于蛋白质组吗?
- 如何选择合适的差异分析R包?
- limma包代码实操
- DEP2-shiny交互界面使用简介
#1. 转录组R包可以用于蛋白质组吗?

转录组差异分析常用R包如limma、DESeq2、edgeR,适用于整数型基因表达计数数据(符合负二项分布)。而蛋白质组数据多为连续型蛋白丰度值(如LFQ强度),常含缺失值和技术变异,通常需经log2变换后采用线性模型方法分析,如limma。因此,必须根据蛋白质组的数据特性选择适配的统计方法。
#2. 如何选择合适的差异分析R包?

差异分析是组学研究的关键环节,直接影响后续结果解读。目前主流蛋白质组分析R包包括:limma、DEP2 和 DEqMS。三者特点对比如下:
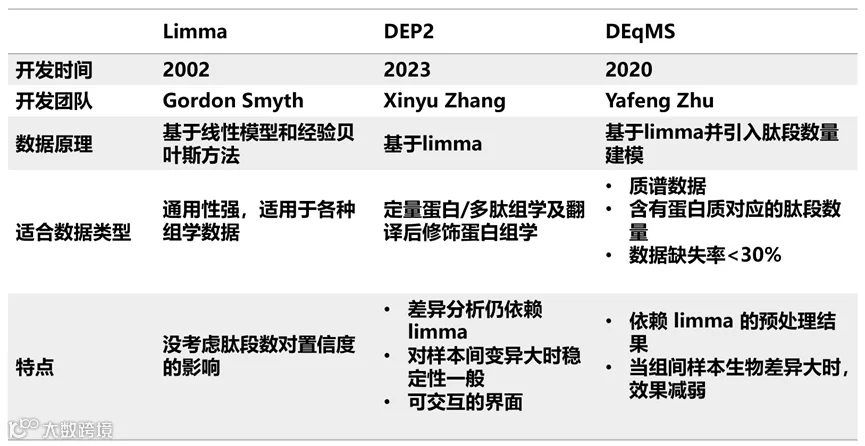
表1. 三种R包应用比较
limma
作为经典差异分析工具,limma基于经验贝叶斯线性模型,广泛应用于微阵列、蛋白芯片及质谱等高通量平台。其优势在于灵活支持数据导入、归一化与建模,适用于多种组学数据类型,通用性强。
DEP2
DEP2(https://github.com/mildpiggy/DEP2)是专为质谱设计的集成化分析工具,兼容蛋白、肽段及翻译后修饰水平数据。该工具整合了差异分析(基于limma)、肽段聚合、可视化及功能富集、PPI网络等下游分析模块,并提供R/Python开发的Shiny交互界面,支持无代码操作,适合初学者快速上手。
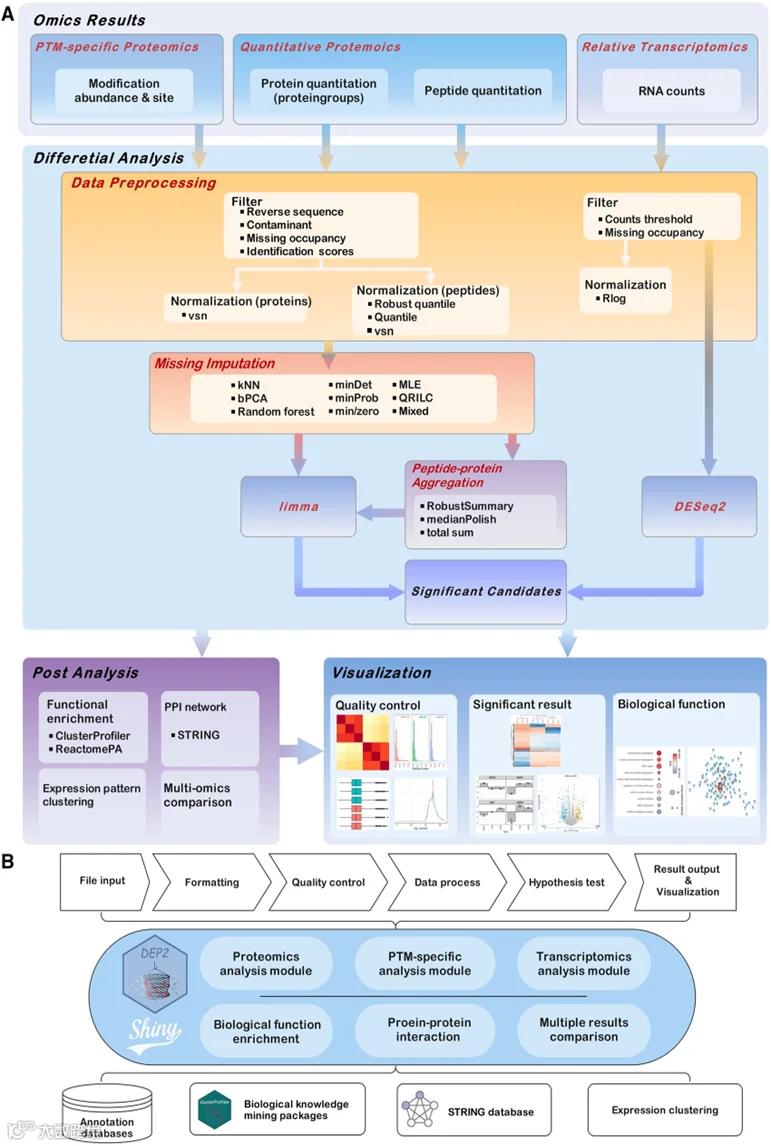
图1. DEP2分析流程
DEqMS
DEqMS(https://github.com/yafeng/DEqM)专为质谱数据优化,在limma基础上引入肽段数量对方差的影响模型,提升低丰度蛋白检出率并降低假阳性。尤其适用于临床样本或磷酸化蛋白组数据。但当样本间生物变异较大时,其性能优势可能减弱。
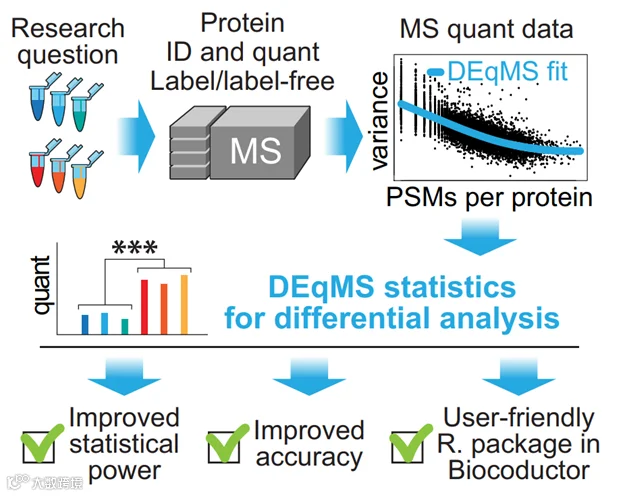
图2. DEqMS包工作流程
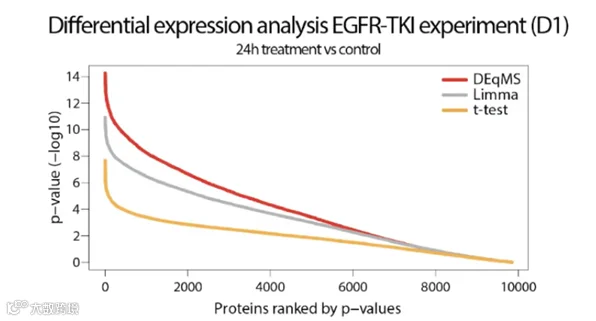
图3. DEqMS方法的灵敏度测试
#3. limma差异分析实操

以下演示基于limma的完整差异分析流程。
数据加载
library(limma)
library(tidyverse)
library(openxlsx)
library(ggpubr)
library(ggthemes)
# 读取数据文件
exp <- read.csv("demo2.csv", row.names = 1) # 第一列作为行名(蛋白质ID)
# 创建样本信息
sample_names <- colnames(exp)
meta <- data.frame(
sample_id = sample_names,
Type = ifelse(grepl("^C", sample_names), "Control", "Treatment"),
stringsAsFactors = FALSE
)
rownames(meta) <- sample_names
# 确保行名和列名一致
identical(rownames(meta), colnames(exp)) # check names数据格式示例:
limma差异分析流程
# 数据预处理:转换为矩阵并进行log2转换(如果数据还没有log转换)
exp_matrix <- as.matrix(exp)
# 检查数据是否需要log转换(通常蛋白质组数据需要log转换)
if(max(exp_matrix, na.rm = TRUE) > 50) {
exp_matrix <- log2(exp_matrix + 1) # 加1避免log(0)
print("数据已进行log2转换")
}
# 设置对比组
meta <- meta %>% mutate(contrast = as.factor(Type))
design <- model.matrix(~ 0 + contrast, data = meta) # 无配对设计
colnames(design) <- c("Control", "Treatment") # 重命名列名
# 拟合线性模型
fit <- lmFit(exp_matrix, design)
# 设置对比矩阵:Treatment vs Control
contrast_matrix <- makeContrasts(
Treatment_vs_Control = Treatment - Control,
levels = design
)
# 进行对比分析
fits <- contrasts.fit(fit, contrast_matrix)
ebFit <- eBayes(fits)
# 获取差异分析结果
limma.res <- topTable(ebFit, coef = "Treatment_vs_Control",
adjust.method = 'fdr', number = Inf)
# 添加蛋白质ID列
limma.res$ID <- rownames(limma.res)
# 处理结果
limma.res <- limma.res %>%
filter(!is.na(P.Value)) %>%
mutate(logP = -log10(P.Value)) %>%
mutate(tag = "Treatment vs Control") %>%
mutate(Gene = ID)
# 设置差异表达标准: FC > 1.5 (log2FC > 0.58), P.value < 0.05
limma.res <- limma.res %>%
mutate(group = case_when(
(P.Value < 0.05 & logFC > 0.58) ~ "up",
(P.Value < 0.05 & logFC < -0.58) ~ "down",
.default = "not sig"
))
# 统计结果
result_summary <- table(limma.res$group)
print("差异表达统计:")
print(result_summary)
# 保存结果
write.xlsx(limma.res, "PRO_Limma_Treatment_vs_Control.xlsx",
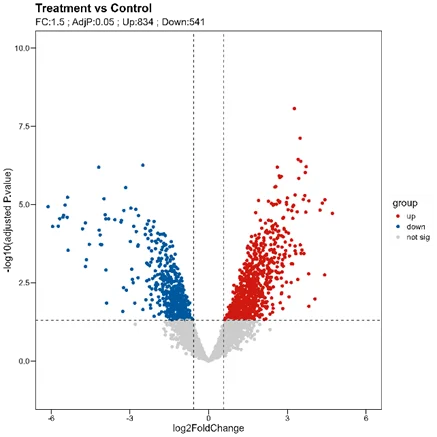
overwrite = TRUE, rowNames = FALSE)绘制火山图
# 可视化
# 准备火山图数据
limma.res <- limma.res %>%
mutate(group = factor(group, levels = c("up", "down", "not sig")))
# 创建标签
my_label <- paste0("FC:1.5 ; AdjP:0.05 ; ",
"Up:", ifelse("up" %in% names(result_summary), result_summary["up"], 0), " ; ",
"Down:", ifelse("down" %in% names(result_summary), result_summary["down"], 0))
# 绘制火山图
p <- ggscatter(limma.res,
x = "logFC", y = "logP",
color = "group", size = 2,
main = "Treatment vs Control",
xlab = "log2FoldChange",
ylab = "-log10(P.value)",
palette = c("#D01910", "#00599F", "#CCCCCC"),
ylim = c(-1, 10),xlim=c(-6,6)) +
theme_base() +
geom_hline(yintercept = -log10(0.05), linetype = "dashed", color = "#222222") +
geom_vline(xintercept = 0.58, linetype = "dashed", color = "#222222") +
geom_vline(xintercept = -0.58, linetype = "dashed", color = "#222222") +
labs(subtitle = my_label) +
theme(plot.background = element_blank())
# 保存图片
ggsave("diff_protein.tiff", p, width = 10, height = 10, dpi = 300)
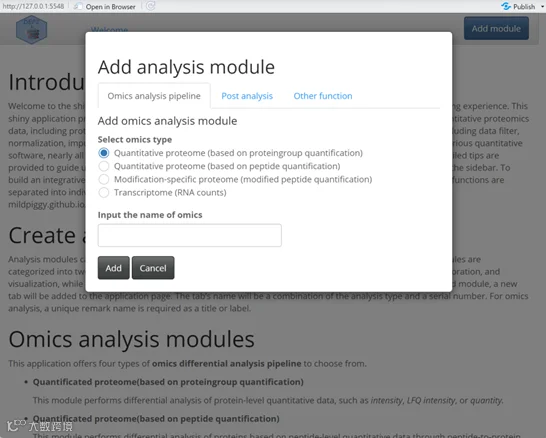
#4. DEP2-shiny交互界面使用

DEP2未收录于Bioconductor官方仓库,需通过GitHub安装,且要求R版本≥4.1.0。
# 从GitHub安装DEP2
if (!requireNamespace("devtools", quietly = TRUE)) {
install.packages("devtools")
}
devtools::install_github("mildpiggy/DEP2")
library("DEP2")
DEP2::run_app()
用户可通过图形界面逐步完成分析,操作直观便捷。
总结与建议
- 初学者希望快速上手且具备图形界面支持,推荐使用DEP2;
- 分析高质量质谱数据且关注低丰度蛋白检出,推荐使用DEqMS(需提供肽段数信息);
- 数据不满足上述条件时,可选用通用性强的limma进行分析。
参考文献:
[1] RITCHIE M E, PHIPSON B, WU D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies [J]. Nucleic Acids Res, 2015, 43(7): e47.
[2] FENG Z, FANG P, ZHENG H, et al. DEP2: an upgraded comprehensive analysis toolkit for quantitative proteomics data [J]. Bioinformatics, 2023, 39(8).
[3] ZHU Y, ORRE L M, ZHOU TRAN Y, et al. DEqMS: A Method for Accurate Variance Estimation in Differential Protein Expression Analysis [J]. Mol Cell Proteomics, 2020, 19(6): 1047-57.